Team:UChicago/Experiments

Methods
Oscillator System
We plan oscillations using western blots and densitometry analysis to track the different phosphorylation states of KaiC. In order to synchronize a population of cells, we used M9 minimal media with no carbon supplements in order to limit the ATP provided to the E.coli cells. This shock was conducted for both one and six hours (See results).
In addition to examining oscillations of our KaiABC biobrick, we also aimed to optimize these oscillations by altering the concentrations of KaiA, which is driven under an L-rhamnose inducible promoter. The input to output ratio of KaiA was first investigated using a 10 fold gradient of L-rhamnose concentrations. We assayed the amount of KaiA produced using Bradford assay and western blots. We estimated the correct concentrations of L-rhamnose as well as the time of induction data from the Paris-Bettencourt 2014 team. Induction was completed for 10 hours. We additionally conducted a time-course of induction for 16 hours in order to determine the saturation point of KaiA, and to examine the degradation rate of KaiA after removal of the inducer from the synchronization/shock in M9 minimal media.
Read-Out System
We plan to assay the read-out system by putting together pMC002 or pMC003 with the KaiC phosphomimetics pMC004-7. pMC004-7 exhibit different phosphorylation states of KaiC, essentially portraying a snapshot of KaiC throughout its oscillations in the KaiABC system. We will pair each of these phosphomimetics with pMC002, containing the master regulator RpaA, its activator SasA, and a response regulator RpaB (the function of this has not been clearly characterized yet). We will also pair each of the phosphomimetics with pMC003, exhibiting the same factors RpaA, RpaB, and SasA with the addition of CikA, a negative regulator of RpaA. We will assay the activation and deactivation of RpaA by measuring the fluorescence of GFP under a plate reader. Given that the LVA-Stop tagged GFP is directly downstream the circadian kaibc promoter, we expect differences in activation/deactivation of RpaA to directly impact the amount of GFP expressed. As such, we expect different fluorescence readings for each of the phosphomimetics. Furthemore, we hypothesize that pMC003 will yield a greater contrast in fluorescence readings between each phosphomimetic, given that it is a negative regulator of RpaA.
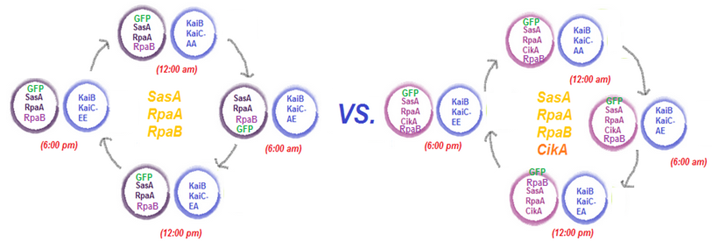
Specific Assays:
Gibson Assembly
Gibson assembly was conducted using NEBs 2x HiFi Assembly Master Mix. Volume of Gibson was halved in order to save reagent. 50 ng of the longest fragment in each reaction was added, and other fragments were added in ratio relative to the longest fragment. 5uL of the master mix was used, and H2O was added to bring whole volume of reaction to 10 uL. Reaction was incubated at 50C for 1 hour.
PCR
Phusion DNAP from Thermo Fisher with HF buffer used for reactions. Each reaction was supplemented with DMSO. Gradient PCRs, miniprep PCRs, and colony PCRs were conducted with 10uL as the final reaction volume. Samples that would be gel extracted were conducted with 50 uL as the final reaction volume. 10 uM primers were added. 30 cycles of PCR were conducted. See Notebook for details on cycle temperatures.
Gel Electrophoresis
Gel set up in TAE buffer. 1% agarose gel used for standard electrophoresis. 0.8% gel used for smaller fragments. 0.7 uL of EtBr added for imaging. Gels run for 30 mins at 120V. Invitrogen 1 kB ladder used for standard protocols.
Gel Extraction
Machery-Nagel gel extraction kit used. See notebook for complete protocol. Samples eluted in 15 uL of elution buffer.
Transformation
Competent cells made by team using RbCl2 and CaCl2 chemically competent protocols.
Efficiencies of competent cells were 4.00 x 10^5 transformants/ng and 1.15 x 10^6 transformants/ng respectively. Transformation was conducted by incubating on ice for 30 mins and conducting a heat shock at 42 C for 1 min. Colonies plated using glass beads method. Incubation at 37C overnight.
Sequencing
Sequencing conducted by the University of Chicago Comprehensive Cancer Center’s sequencing centers.
Growing cultures
Cultures grown for induction were incubated at 37C. OD600 for initial assays were measured to be around 1.5. In latter assays, cells were diluted to be OD 600 ~0.6-0.7. Initial cultures were always grown in LB.
Induction
Induction of rhamnose to drive KaiA expression conducted in M9 minimal media with supplements: 0.4% glycerol and 0.1% casamino acids. Induction solutions made with varying concentrations of rhamnose in order to characterize the input-output ratio of rhamnose to KaiA. This was significant for us to be able to vary the concentrations of KaiA and to develop more robust oscillations. Induction conducted for 10 hours at 30C. Induction protocol followed from Silver Lab (Chen, A. H., Lubkowicz, D., Yeong, V., Chang, R. L., & Silver, P. a. (2015a). Transplantability of a circadian clock to a noncircadian organism. Science Advance, 1(5), 1–6. http://doi.org/10.1126/sciadv.1500358) .
Synchronization
Synchronization to synchronize the KaiABC clock was conducted by incubating induced cells in M9 minimal media only (no supplements or inducer). Synchronization conducted for both 1 hour and 6 hours. Silver lab reported 1 hour synchronization while Rust lab has seen 6 hours to be adequate for cyanobacteria.
Oscillation Time Course
Following synchronization, cells were reintroduced in M9 media with 1mM leucine and 0.5% succinate supplements to allow for slow growth. Oscillations were tracked by freezing 2 mL cell samples every 6 hours in a -80C freezer. The time duration for tracking oscillations was set at 3 days. No rhamnose was added back, due to Silver lab’s reports of desynchronization of oscillations attributed to reintroduction of inducer with their construct.
Western Blot
In order to track expression of KaiA,B,C, and phosphorylation states of KaiC western blots were conducted. For simple expression assays, such as when characterizing the pRha promoter and quantifying KaiA expression, a 14-20% BioRad precast protein gel was used during SDS page gel run. For tracking different phoshphorylation states of KaiC, a 4-7% BioRad precast protein gel was used. Bradford assays were conducted before SDS-page in order to standardize the amount of protein added per sample. A positive control of cyanobacterial lysate, and a negative control of E.coli cells containing an empty Cam circular backbone were run. Purified protein samples provided by the Rust lab were run to quantify amount of KaiA, B, and C expressed by transformed E.coli cells. Transfer accomplished using PVDF membranes. Membrane sandwich apparatus was prepared under Towbin transfer buffer. Blocking to prevent non-specific binding of antibodies was conducted using TBST+2% dry milk. This was followed by a stain of primary anti-rabbit antibodies for the Kai proteins provided by the Rust Lab. The membranes were washed using wash buffer then stained with anti-goat HRP secondary antibody allowing us to visualize the primary antibody stain. The membranes were washed then imaged using chemilumenescnece imaging systems.
Imaging and Quantification
Densitometry of images was conducted in ImageJ. For the induction time course and the oscillation assays (oscillation data not up on wiki, but to be presented at the Jamboree), the nanogram amount of each protein was determined by calibrating to a logarithm curve of purified standards of known concentration ran on the same Western blot. This allowed for determination of accurate stoichiometric ratios between KaiA on the rhamnose inducible promoter and KaiB and KaiC, both on the constitutive promoter. Normalization for OD was performed for post-shock time points, including oscillation time points, to control for changes in cell density and protein degradation after induction of KaiA was turned off.
Like our team Facebook page, Genehackers@UChicago!
Questions? Comments? Send us an email!