Team:DTU-Denmark/Project/Background

The Synthesizer
Figure 1. The Syntesizer platform.
Nonribosomal petides (NRP) are a diverse class of short, bioactive peptides isolated from plant, fungi, and bacteria. Clinical uses of NRPs include antibiotics, immunosuppressants, antifungal, and antitumor drugs [1]. They are synthesized by highly modular enzymes called nonribosomal peptide synthetases (NRPS). Through repeated rounds of condensations of more than 500 different amino acids in an assembly line manner, they synthesize a range of different peptides with diverse bioactivities [2]. Discovery of novel NRPs may lead to development of novel drugs, but is limited by development cost and time of drug discovery [3,4]. Some existing nonribosomal peptides on the market are associated with severe side-effects [5] or reduced activity due to resistance [6], while the implementation of other potential NRP drugs are limited due to their toxicity [7]. Improvement of existing NRP drugs, an area which is largely unexploited due to the complexity of the synthetases, has great potential for pharmaceutical improvement. The DTU Synthesizer Team built a novel platform for generating improved NRP drug libraries. The platform in Bacillus subtilis combines bioinformatics and implementation of advanced multiplex genetic tools to generate drug libraries using recombineering with short single-stranded DNA.
Introduction
Unlike proteins, nonribosomal peptides are not synthesized through translation, but rather through sequential condensation of amino acids by large multimodular enzymes, called nonribosomal peptide synthetases (NRPS)[1,8]. It is this complex pattern of condensation of amino acids that attributes to the biological activity of nonribosomal peptides (NRP). NRP synthetases do not rely on an external template for synthesis of their products. They can be divided up into modules that are each responsible for incorporating one additional amino acid onto the growing peptide chain, much like an assembly line (Figure 2) [9]. At the end of the assembly line, the growing peptide is released as a linear or cyclic peptide.

Figure 2. NRPS are highly modular. Tyrocidine Synthetase (tycA-C) consists of ten modules, two epimerization domains, and a thioesterase (TE) domain. Tyrocidine precursors are assembled one-by-one in an assembly line manner. The TE domain cyclizes the tyrocidine upon releasing it from the NRPS.
Each module, with the exception of the initiation module, consists of at least three domains. These domains are responsible for activating the monomer (adenylation (A) domain), holding the activated monomer (peptidyl carrier protein (PCP) domain), and amino acid condensation (condensation (C) domain). The C-domain is lacking in the initiation module and the terminal module contains an additional required domain responsible for termination and release of the product called the thioesterase (TE) domain [8].
Adenylation domains
The adenylation domains are responsible for activating and attaching the amino acid monomers to the PCP domain. They act as gatekeepers, ensuring that only the desired monomers are incorporated. They may be highly specific and only bind a single amino acid, or as in the case with Tyrocidine, (see below) allow multiple similar amino acids to be incorporated in the peptide [2].
Peptidyl carrier protein domains
The monomers are covalently bound to the PCP domains through thioester bonds. Before they can accept the monomers, they must be activated by posttranslational modification. A 4'-phosphopantetheinyl transferases (PPTase) transfers a 4'-phosphopantetheine (4'-PP) moiety, carrying the sulfhydryl group required for thioester bond formation, from coenzyme A to a conserved serine residue in the PCP domain [8].
Condensation domains
Elongation of the peptidyl chain is performed by the C domains, which catalyzes the condensation of the peptidyl chain bound to the upstream PCP domain and the amino acid bound to the downstream PCP domain. These domains have strong stereoselectivity and may have some specificity towards the side chain of the amino acid incorporated by the A domain of the same module, whereas little specificity has been observed towards the peptidyl chain [10].
Thioesterase domains
The TE domain catalyzes the final step of NRP synthesis. It is the TE domain that determines if the peptide is released or cyclized to form a cyclic structure. Despite, the important role of TE-domains they are the least understood in the pathway. While it is possible to predict A domain specificity, it is not possible to predict TE affinity [11]. The tyrocidine TE domain (Figure 2) shows high flexibility in the number of different substrates it will accept for cyclization.
Engineering of NRPS
With a pool of more than 500 different amino acid monomers, a peptide of only nine monomers have almost infinite possibilities of composition. Yet, to our knowledge, improvement of existing NRP drugs through genomic engineering of NRPS in vivo still remains unaccomplished. This is somewhat surprising as it has been possible to predict NRP products based on bioinformatics analysis of NRPS clusters for quite some time [11-13]. Two years ago, the iGEM team from Heidelberg tried to advance synthesis of synthetic peptides by combining modules from different NRPS to create potentially any desired peptide. In addition, they expressed an NRPS module (indC), which leads to the formation of a blue color after the NRP is released from the thioesterase module. When placed after an NRPS module, it was possible to produce tagged peptides. Such a tagging system would greatly improve the synthesis of synthetic NRPS-derived peptides, but to our knowledge, it has not been possible to repeat the experiment within the group at Heidelberg. This illustrates the complexity of NRPS engineering, despite its modularity. Crystallization and structural studies have highlighted the specific and evolutionary evolved interaction between modules as crucial for NRPS function [14][15].
| Domain | Position (Stachelhaus code) | Similarity | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 235 | 236 | 239 | 278 | 299 | 302 | 322 | 330 | 331 | 317 | ||
| Aad | E | P | R | N | I | V | E | F | V | K | 94% |
| Ala | D | L | L | F | G | I | A | V | L | K | 55% |
| Asn | D | L | T | K | L | G | E | V | G | K | 90% |
| Asp | D | L | T | K | V | G | H | I | G | K | 100% |
| Cys(1) | D | H | E | S | D | V | G | I | T | K | 96% |
| Cys(2) | D | L | Y | N | L | S | L | I | W | K | 88% |
| Dab | D | L | E | H | N | T | T | V | S | K | 100% |
| Dhb/Sal | P | L | P | A | Q | G | V | V | N | K | 83% |
| Gln | D | A | Q | D | L | G | V | V | D | K | 100% |
| Glu(1) | D | A | W | H | F | G | G | V | D | K | 95% |
| Glu(2) | D | A | K | D | L | G | V | V | D | K | 95% |
| Ile(1) | D | G | F | F | L | G | V | V | Y | K | 92% |
| Ile(2) | D | A | F | F | Y | G | I | T | F | K | 100% |
| Leu(1) | D | A | W | F | L | G | N | V | V | K | 99% |
| Leu(2) | D | A | W | L | Y | G | A | V | M | K | 100% |
| Leu(3) | D | G | A | Y | T | G | E | V | V | K | 100% |
| Leu(4) | D | A | F | M | L | G | M | V | F | K | 97% |
| Orn(1) | D | M | E | N | L | G | L | I | N | K | 100% |
| Orn(2) | D | V | G | E | I | G | S | I | D | K | 98% |
| Phe | D | A | W | T | I | A | A | V | C | K | 88% |
| Phg/hPhg | D | I | F | L | L | G | L | L | C | K | 80% |
| Pip/Pip-@ | D | F | Q | L | L | G | V | A | V | K | 75% |
| Pro | D | V | Q | L | I | A | H | V | V | K | 87% |
| Ser | D | V | W | H | L | S | L | I | D | K | 90% |
| Thr/Dht | D | F | W | N | I | G | M | V | H | K | 91% |
| Tyr(1) | D | G | T | I | T | A | E | V | A | K | 100% |
| Tyr(2) | D | A | L | V | T | G | A | V | V | K | 80% |
| Tyr(3) | D | A | S | T | V | A | A | V | C | K | 78% |
| Val(1) | D | A | F | W | I | G | G | T | F | K | 96% |
| Val(2) | D | F | E | S | T | A | A | V | Y | K | 94% |
| Val(3) | D | A | W | M | F | A | A | V | L | K | 95% |
| Variability | 3% | 16% | 16% | 39% | 52% | 13% | 26% | 23% | 26% | 0% | |
| Table 1. Stachelhaus code showing position in Stachelhaus code (as defined by multiple alignment) with A domain specificity [16].The data consists of 160 A-domains and is adapted after Stachelhaus et al. 2009. | |||||||||||
Prediction of adenylation domain specificity
antiSMASH, currently the leading method for prediction of NRPS products, predicts a consensus product from an NRPS cluster based on three different methods: NRPSpredictor2, Stachelhaus, and Minova et al. [11,17]. Stachelhaus et al. aligned the binding pocket of 160 adenylation domains based on the first solved structure of NRPS A domain, PheA using a structural alignment approach[15]. By trimming off the sequence, 10 core amino acids encoding specificity in the binding pocket were identified, giving rise to the Stachelhaus code, see Table 1. In addition, it was shown that by modifying the binding pocket in silico, substrate affinity could be altered or relaxed [16].
Prediction of substrate affinity is sometimes complicated by the fact that adenylation domains may incorporate a variety of different amino acids. This is represented by a redundancy in the Stachelhaus code, though this is very low[16]. Tyrocidine is one example of this phenomenon. Tyrocidine is a commercially available mixture of non-ribosomal antibiotics synthesized by Brevibacillus parabrevis. It consists of four decapeptides varying at three amino acids (see Table 2) and is synthetized by the NRPS Tyrocidine Synthetase A-C, containing 1, 3, and 6 modules, respectively. (see Figure 2)
Tyrocidine has a unique mode of action wherein it disrupts the function of the cell membrane. Unfortunately, it has high toxicity towards human blood and reproductive cells and can only be used in topical applications. This makes tyrocidine an interesting target for drug improvement. Our initial plan was to express tyrocidine in Bacillus subtilis, but due to time constraints we decided to focus on manipulating the native Surfactin synthetase with our oligo recombineering technology.
|
||||
|---|---|---|---|---|
|
|
Amino acid position |
|||
|
Tyrocidine |
3 |
4 |
7 |
|
|
A |
L-Phe |
D-Phe |
L-Tyr |
|
|
B |
L-Trp |
D-Phe |
L-Tyr |
|
|
C |
L-Trp |
D-Trp |
L-Tyr |
|
|
D |
L-Trp |
D-Trp |
L-Trp |
|
Targetted engineering of adenylation domains
Potential NRP targets for drug improvement
In addition to tyrocidine, ciclosporin (or cyclosporine) is an important nonribosomal peptide drug used as an immunosuppressant in transplantations[5]. It consists of eleven amino acids, which are cyclized upon release from the NRPS. Like tyrocidine, ciclosporin contains D-amino acids as well as amino acids with modifications (Figure #). It was first isolated from the filamentous fungi Tolypocladium inflatum in 1969. In 1972, its function as an immunosuppresant was discovered by Sandoz (now Novartis). Despite, its wide use in clinical applications, it is associated with side effects [5]. In cyclosporine G, which is also isolated from Tolypocladium inflatum, the a-aminobutyric acid residue in position 2 has been replaced by norvaline [18]. Ciclosporin G shows reduced side effects in some clinical applications and it has demonstrated potential improvements in immunosuppressant treatments. A total of 25 derivatives of ciclosporin are known [19]. Comparing the number of known derivatives to the actual potential diversity in compounds of an 11-mer cyclic peptide (11500) it is clear that only a miniscule fraction of potential compounds have been tested. Even substitution of a single amino acid would yield more than 5,000 potential drugs that could be screened for improved function.
Hypothesis: Targetted engineering of adenylation domains using MAGE in Bacillus
As highlighted above, there are multiple potential NRPS candidates that can be used for drug improvement through screening of NRP product derivatives. Since the 1990’ies, the Stachelhaus code has made it possible to target specific residues of the adenylation domain to create this variability in products, yet limitations in genetic tools for editing of these large proteins with a size approximately equivalent to the large subunit of the prokaryotic ribosome. Even though the changes required to potentially alter the specificity of the A domain is only around 1-10 amino acids, according to the Stachelhaus code, transformations require introduction of selection cassettes. As each NRPS module is ~1,500 amino acids and often multiple modules are encoded in one open reading frame, even a few minor modifications require assembly and introduction of large expression cassettes and multiple markers. For example, the NRPS responsible for cyclosporine synthesis in Tolypocladium inflatum is encoded in a single CDS of 45.8 kb [19]. Amplification of 45.8 kb nucleotides is not feasible with standard PCR techniques and would be considerably expensive to synthesize by cheap error-prone DNA synthesis methods.
We hypothesized that NRPS directed evolution targeting using the Stachelhaus code could lead to improvement of NRP drugs. We proposed that the recent advantage in oligo mediated recombineering (MAGE) using short single-stranded DNA (ssDNA) can be applied to generate this diversity.
Oligo mediates recombineering
Recombination-mediated genetic engineering or recombineering utilizes homologous recombination to facilitate genetic modifications at any desired target by flanking the mutated sequence with homologous regions. One system for recombineering in E. coli is the λ phage derived λ Red, consisting of the genes encoding three proteins, Gam, Exo and Beta. Gam prevents degradation of linear dsDNA by inhibition of nucleases, Exo degrades dsDNA in a 5'-3' direction yielding ssDNA, and Beta facilitates recombination by binding to the ssDNA [20].
Multiplex Automated Genome Engineering (MAGE)
Wang et al. developed a method for rapid and efficient targeted evolution of cells through cyclical recombineering with ssDNA (oligos) in E. coli. Using this automated method, they showed more than five-fold improved lycoprene production in E. coli in three days [21]. Through MAGE it is possible to simultaneously target many different loci or target the same locus with a pool of multiple and/or degenerate oligos. By using multiple oligos targetting the same locus, it is possible to generate a library of mutants varying only at the target locus in a short amount of time. The genetic variation in the population of cells will be a function of the degenerate pool complexity and combinatorial arrangements of the modifications at different loci [21,22].
 The MAGE protocol utilizes the λ Red recombination system in combination with a temporary inactivation of the mismatch repair system [22]. As MAGE utilizes oligos, only the Beta protein of the λ Red system is required. When E. coli is targeted with single-stranded oligos, Beta stabilizes them inside the cell and facilitates homologous recombination into the genome, see Figure 3. Interestingly, this targeting is many-fold more effective, if the oligo is targeting the lagging strand compared to the leading strand [20,21,23,24]. Based on this observation, it is proposed that Beta functions through binding to the lagging strand of the replication fork and by stabilizing oligo integration between discontinuous Okazaki fragments [20,24].
The MAGE protocol utilizes the λ Red recombination system in combination with a temporary inactivation of the mismatch repair system [22]. As MAGE utilizes oligos, only the Beta protein of the λ Red system is required. When E. coli is targeted with single-stranded oligos, Beta stabilizes them inside the cell and facilitates homologous recombination into the genome, see Figure 3. Interestingly, this targeting is many-fold more effective, if the oligo is targeting the lagging strand compared to the leading strand [20,21,23,24]. Based on this observation, it is proposed that Beta functions through binding to the lagging strand of the replication fork and by stabilizing oligo integration between discontinuous Okazaki fragments [20,24].
Figure 3. Proposed mechanism for function of Beta in oligo recombineering during DNA replication. Beta stabilises oligo that is incorporated between discontinious Okazaki fragments on the lagging strand [23]. From Carr et al. 2014.
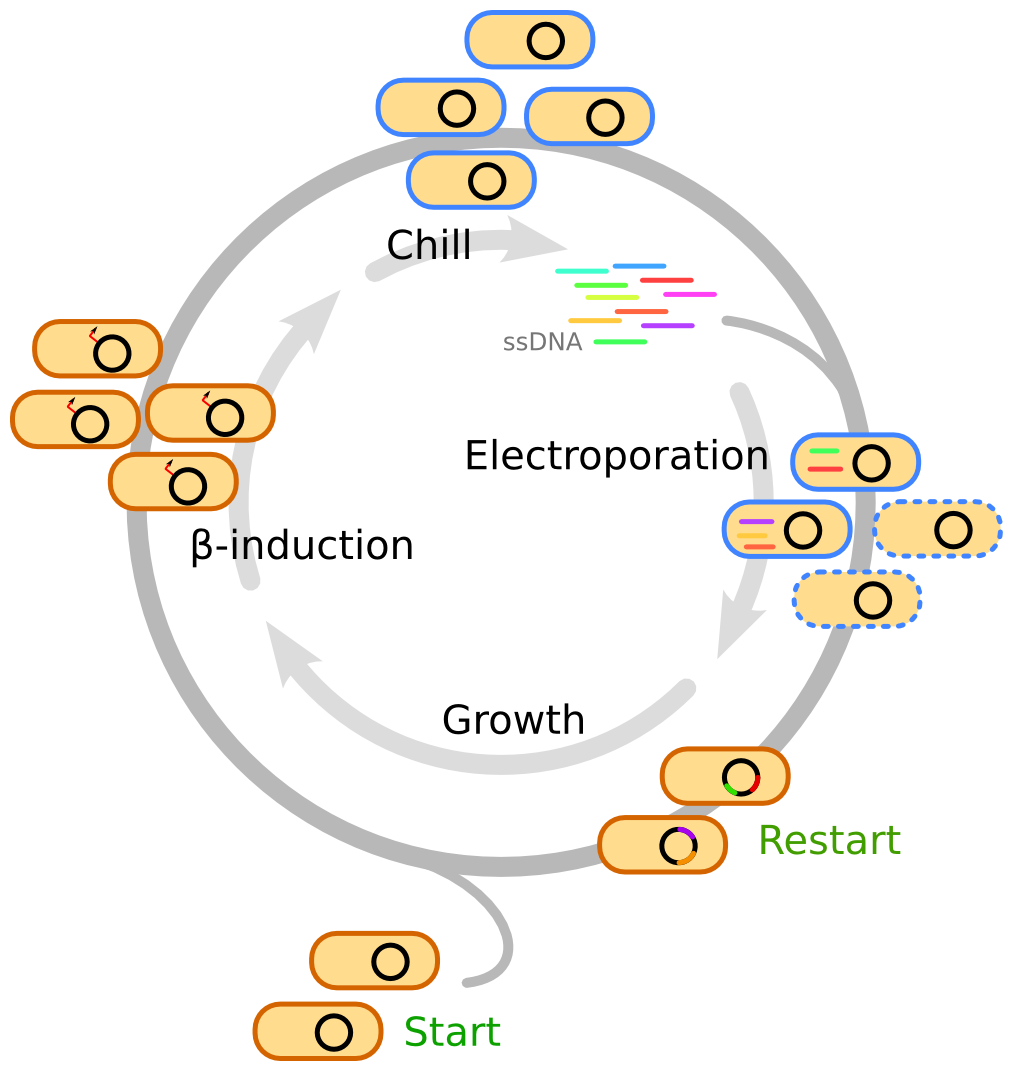 MAGE becomes multiplex and automated, when the oligo recombineering steps are repeated multiple times. In short, the cells are first grown to mid-log phase, followed by induction of Beta. The cells are then chilled to 4°C and washed in multiple steps to improve their competence. The oligos are added to the cells which are then electroporated. The cycle is repeated every 2-3 hours allowing the cells to recover in between rounds of electroporation, see Figure4 [22].
MAGE becomes multiplex and automated, when the oligo recombineering steps are repeated multiple times. In short, the cells are first grown to mid-log phase, followed by induction of Beta. The cells are then chilled to 4°C and washed in multiple steps to improve their competence. The oligos are added to the cells which are then electroporated. The cycle is repeated every 2-3 hours allowing the cells to recover in between rounds of electroporation, see Figure4 [22].
Figure 4. Protocol for MAGE induction. Repeated rounds of electroporation with ssDNA pool, followed by recovering period, induction of Beta, and preparations of cells for next round of MAGE. Credit: Michael Schantz Klausen.
Chip-based oligonucleotide synthesis
Traditional column-based oligo synthesis is costly for large scale MAGE experiments. Synthesis of 1,000 90-mer column-based oligos costs approximately $36,000 USD [25]. An alternative to this method is the use of Microchip DNA arrays. Microchips become advantageous when the number of oligos increases. With Microchip DNA arrays it is possible to synthesize up to 12,472 different 130-mer oligos for 2,000 USD and up to 92,918 oligos for 5,000 USD [26].
 The oligos are synthesized with two 20 nucleotide flanking sequences (barcodes). These barcodes must include a thymidine immediately upstream and a DpnII restriction site immediately downstream of the oligo, while the rest of the barcodes can be designed for amplification with a specific primer. By using different barcodes, it is possible to design one oligo chip for multiple MAGE experiments. The thymidine allows amplification with an uracil-containing primer. In these primers U is substituted for T in the primer. The barcode can then be excised from the oligo using USER enzyme, DnpII, and a guide primer[25]. Despite the low cost of this method, it comes at a price: Oligos are delivered as a single mix of all the oligos and they are each in the picomolar range. This requires extra processing and amplification steps before the oligos can be used in a large-scale MAGE experiment.
The oligos are synthesized with two 20 nucleotide flanking sequences (barcodes). These barcodes must include a thymidine immediately upstream and a DpnII restriction site immediately downstream of the oligo, while the rest of the barcodes can be designed for amplification with a specific primer. By using different barcodes, it is possible to design one oligo chip for multiple MAGE experiments. The thymidine allows amplification with an uracil-containing primer. In these primers U is substituted for T in the primer. The barcode can then be excised from the oligo using USER enzyme, DnpII, and a guide primer[25]. Despite the low cost of this method, it comes at a price: Oligos are delivered as a single mix of all the oligos and they are each in the picomolar range. This requires extra processing and amplification steps before the oligos can be used in a large-scale MAGE experiment.
Figure 5. Schematic represenation of MO-MAGE Oligo Processing.
Oligo design
While the recombination frequency of MAGE can be increased by doing multiple cycles, care should be taken when designing oligos to ensure high efficiency for each individual cycle, see Figure 5. There are several parameters that can be optimized to increase the recombination frequency: The oligos should target the lagging strand of the replication fork, as this improves efficiencies by 10-100 fold. In addition, the folding energy of the oligo should be considered, as it may form hairpins if it is too low, preventing incorporation. The frequency is also dependent on the length of the oligo, as shorter oligos are less efficient due to their lower hybridization energy to the chromosome, while longer oligos have a higher tendency to form hairpins. 70-90-mer oligos seem to be the most efficient in E. coli [22].
Designing many optimized oligos for MAGE experiments is a time consuming task. Considering a 90-mer oligo with a single mismatch and 15nt homology arms results in 60 possible oligos all with different secondary structures and consequently different recombineering efficiencies. Much of the time spent on designing oligos can be saved by using tools such as MODEST [27].
Oligo mediates recombineering
The described λ Red recombineering system has been exploited in E. coli over the last few years, but the technology has not been adopted for genome editing to the same extent in other microorganisms. Application of λ Red recombineering systems has been described in Bacillus subtilis [-1] Instead of 90-mer oligos, long single stranded DNA fragments of approximately 2,000 nucleotides, generated by PCR, were used [28]. Besides expression of Beta, Sun et al. also expressed homologous recombinases from other phages. The gene product of region 35 (GP35) from the native B. subtilis phage SPP1 is homologous to Beta. Gene product of region 34 (GP34) encodes an endonuclease similar to λ Exo and GP36 is an ssDNA binding protein [29,30]. This native Bacilli reecombineering system yielded higher efficiency compared to λ Red in B. subtilis, but lower efficiency in E. coli[28].
Based on heterologous expression of many recombinases of different origin in Bacillus subtilis, it was concluded that recombineering efficiencies were optimized by using a recombinase derived from a phage, whose host was closely related to the MAGE host. We noticed that codon usage of lambda beta is not optimal for expression of B. subtilis and that expression level (likely due to codon usage) varied among the recombinases tested in the study. In addition, the length of the oligo they tested is drastically different from the optimal length in E. coli based on the λ Red Lambda recombineering system and their engineering was not done in multiplex mode.
Our first goal was therefore to generate a MAGE compatible Bacillus subtilis strain that could be transformed with short oligos.
References
- Walsh, C. T. (2008). The Chemical Versatility of Natural-Product Assembly Lines. Acc. Chem. Res., 41(1), 4–10. doi:10.1021/ar7000414
- Caboche, S., Leclere, V., Pupin, M., Kucherov, G., & Jacques, P. (2010). Diversity of Monomers in Nonribosomal Peptides: towards the Prediction of Origin and Biological Activity. Journal of Bacteriology, 192(19), 5143–5150. doi:10.1128/jb.00315-10
- DiMasi, J. A., Hansen, R. W., & Grabowski, H. G. (2003). The price of innovation: new estimates of drug development costs. Journal of Health Economics, 22(2), 151–185. doi:10.1016/s0167-6296(02)00126-1
- DiMasi, J. A., Grabowski, H. G., & Hansen, R. W. (2015). The Cost of Drug Development. N Engl J Med, 372(20), 1972–1972. doi:10.1056/nejmc1504317
- Lee, J.-H. (2010). Use of Antioxidants to Prevent Cyclosporine A Toxicity. Toxicological Research, 26(3), 163–170. doi:10.5487/tr.2010.26.3.163
- Henderson, J. C., Fage, C. D., Cannon, J. R., Brodbelt, J. S., Keatinge-Clay, A. T., & Trent, M. S. (2014). Antimicrobial Peptide Resistance of Vibrio cholerae Results from an LPS Modification Pathway Related to Nonribosomal Peptide Synthetases . ACS Chemical Biology, 9(10), 2382–2392. doi:10.1021/cb500438x
- Kohli, R. M., Walsh, C. T., & Burkart, M. D. (2002). Biomimetic synthesis and optimization of cyclic peptide antibiotics. Nature, 418(6898), 658–661. doi:10.1038/nature00907
- Finking, R., & Marahiel, M. A. (2004). Biosynthesis of Nonribosomal Peptides 1 . Annu. Rev. Microbiol., 58(1), 453–488. doi:10.1146/annurev.micro.58.030603.123615
- Strieker, M., Tanović, A., & Marahiel, M. A. (2010). Nonribosomal peptide synthetases: structures and dynamics. Current Opinion in Structural Biology, 20(2), 234–240. doi:10.1016/j.sbi.2010.01.009
- Lautru, S. (2004). Substrate recognition by nonribosomal peptide synthetase multi-enzymes. Microbiology, 150(6), 1629–1636. doi:10.1099/mic.0.26837-0
- Blin, K., Medema, M. H., Kazempour, D., Fischbach, M. A., Breitling, R., Takano, E., & Weber, T. (2013). antiSMASH 2.0--a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Research, 41(W1), W204–W212. doi:10.1093/nar/gkt449
- Medema, M. H., Blin, K., Cimermancic, P., de Jager, V., Zakrzewski, P., Fischbach, M. A., … Breitling, R. (2011). antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Research, 39(suppl), W339–W346. doi:10.1093/nar/gkr466
- Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H. U., Bruccoleri, R., … Medema, M. H. (2015). antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Research, 43(W1), W237–W243. doi:10.1093/nar/gkv437
- Sundlov, J. A., Shi, C., Wilson, D. J., Aldrich, C. C., & Gulick, A. M. (2012). Structural and Functional Investigation of the Intermolecular Interaction between NRPS Adenylation and Carrier Protein Domains. Chemistry & Biology, 19(2), 188–198. doi:10.1016/j.chembiol.2011.11.013
- Conti, E. (1997). Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. The EMBO Journal, 16(14), 4174–4183. doi:10.1093/emboj/16.14.4174
- Stachelhaus, T., Mootz, H. D., & Marahiel, M. A. (1999). The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chemistry & Biology, 6(8), 493–505. doi:10.1016/s1074-5521(99)80082-9
- Minowa, Y., Araki, M., & Kanehisa, M. (2007). Comprehensive Analysis of Distinctive Polyketide and Nonribosomal Peptide Structural Motifs Encoded in Microbial Genomes. Journal of Molecular Biology, 368(5), 1500–1517. doi:10.1016/j.jmb.2007.02.099
- CALNE, R. (1985). CYCLOSPORIN G: IMMUNOSUPPRESSIVE EFFECT IN DOGS WITH RENAL ALLOGRAFTS. The Lancet, 325(8441), 1342. doi:10.1016/s0140-6736(85)92844-2
- Bushley, K. E., Raja, R., Jaiswal, P., Cumbie, J. S., Nonogaki, M., Boyd, A. E., … Spatafora, J. W. (2013). The Genome of Tolypocladium inflatum: Evolution, Organization, and Expression of the Cyclosporin Biosynthetic Gene Cluster. PLoS Genet, 9(6), e1003496. doi:10.1371/journal.pgen.1003496
- Mosberg, J. A., Lajoie, M. J., & Church, G. M. (2010). Lambda Red Recombineering in Escherichia coli Occurs Through a Fully Single-Stranded Intermediate. Genetics, 186(3), 791–799. doi:10.1534/genetics.110.120782
- Wang, H. H., Isaacs, F. J., Carr, P. A., Sun, Z. Z., Xu, G., Forest, C. R., & Church, G. M. (2009). Programming cells by multiplex genome engineering and accelerated evolution. Nature, 460(7257), 894–898. doi:10.1038/nature08187
- Wang, H. H., & Church, G. M. (2011). Multiplexed Genome Engineering and Genotyping Methods. Synthetic Biology, Part B - Computer Aided Design and DNA Assembly, 409–426. doi:10.1016/b978-0-12-385120-8.00018-8
- Carr, P. A., Wang, H. H., Sterling, B., Isaacs, F. J., Lajoie, M. J., Xu, G., … Jacobson, J. M. (2012). Enhanced multiplex genome engineering through co-operative oligonucleotide co-selection. Nucleic Acids Research, 40(17). doi:10.1093/nar/gks455
- Gallagher, R. R., Li, Z., Lewis, A. O., & Isaacs, F. J. (2014). Rapid editing and evolution of bacterial genomes using libraries of synthetic DNA. Nature Protocols, 9(10), 2301–2316. doi:10.1038/nprot.2014.082
- Bonde, M. T., Kosuri, S., Genee, H. J., Sarup-Lytzen, K., Church, G. M., Sommer, M. O. A., & Wang, H. H. (2015). Direct Mutagenesis of Thousands of Genomic Targets Using Microarray-Derived Oligonucleotides. ACS Synthetic Biology, 4(1), 17–22. doi:10.1021/sb5001565
- http://customarrayinc.com/oligos_main.htm (Link and numbers confirmed at wikifreeze 09/19.15)
- Bonde, M. T., Klausen, M. S., Anderson, M. V., Wallin, A. I. N., Wang, H. H., & Sommer, M. O. A. (2014). MODEST: a web-based design tool for oligonucleotide-mediated genome engineering and recombineering. Nucleic Acids Research, 42(W1), W408–W415. doi:10.1093/nar/gku428
- Sun, Z., Deng, A., Hu, T., Wu, J., Sun, Q., Bai, H., … Wen, T. (2015). A high-efficiency recombineering system with PCR-based ssDNA in Bacillus subtilis mediated by the native phage recombinase GP35. Applied Microbiology and Biotechnology, 99(12), 5151–5162. doi:10.1007/s00253-015-6485-5
- Vellani, T. S., & Myers, R. S. (2003). Bacteriophage SPP1 Chu Is an Alkaline Exonuclease in the SynExo Family of Viral Two-Component Recombinases. Journal of Bacteriology, 185(8), 2465–2474. doi:10.1128/jb.185.8.2465-2474.2003
- Seco, E. M., Zinder, J. C., Manhart, C. M., Lo Piano, A., McHenry, C. S., & Ayora, S. (2012). Bacteriophage SPP1 DNA replication strategies promote viral and disable host replication in vitro. Nucleic Acids Research, 41(3), 1711–1721. doi:10.1093/nar/gks1290

Department of Systems Biology
Søltofts Plads 221
2800 Kgs. Lyngby
Denmark
P: +45 45 25 25 25
M: dtu-igem-2015@googlegroups.com








