Team:DTU-Denmark/Project/MAGE

Overview Strain
In order for MAGE (Multiplex Automated Genome Engineering) to work at a high efficiency, a strain with inserted recombinase and inhibited or knocked out mismatch repair gene has to be used [1]. In our project, two different recombinases were used: a recombination protein Beta from the E. coli phage Lambda, which was codon optimized for B. subtilis 168, and GP35, a recombinase from the B.subtilis phage SPP1 [2]. The mismatch repair proteins known as MutS and MutL were knocked out by transforming pSB1C3_recombinase plasmid into the B. subtilis W168. Since the MutL protein is dependent on the binding of MutS, the knockout of the mutS disables the function of the MutL protein [3]. For doing proof of concept of MAGE in B. subtilis 168 four different strains was produced via genetic recombineering. All derived from Bacillus subtilis 168 WT:
- ∆amyE::beta-neoR
- ∆amyE::GP35-neoR
- ∆mutS::beta-neoR
- ∆mutS::GP35-neoR
Background Strain
Methods Strain
All the strain were made by homologous recombineering. Plasmids containing cassettes that were able to do a double-crossover homologous recombineering into the genome of B. subtilis 168 (referred to as knockout (KO) plasmid). These were used to, simultaneously, delete the desired gene (amyE or mutS) and inserting the expression cassette for one of the recombinases: beta or GP35.
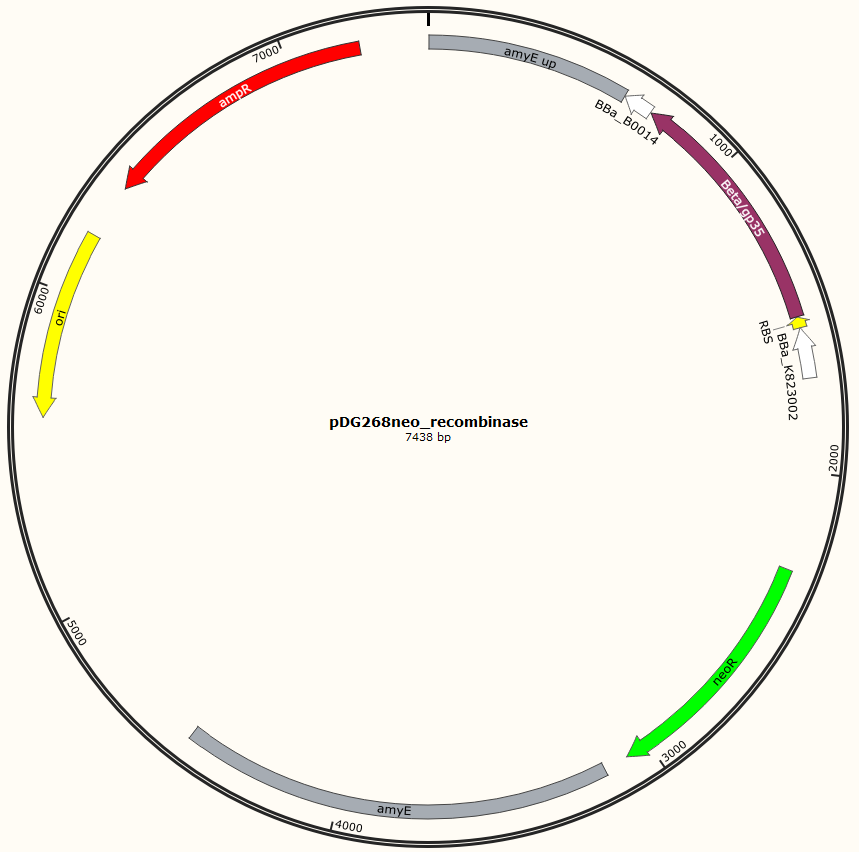
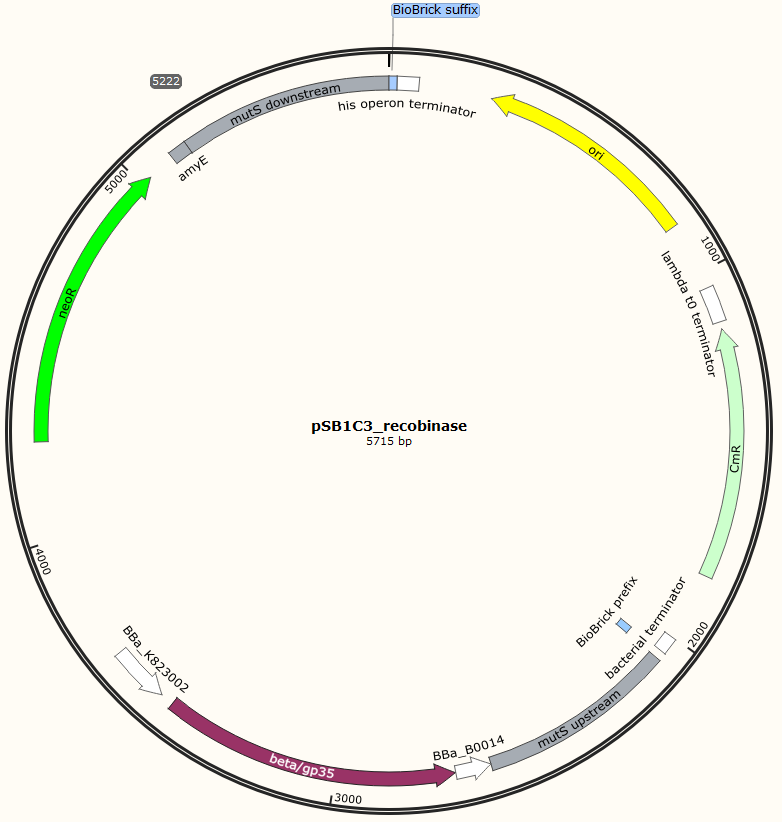
Four different plasmids was assembled to make the four MAGE ready strains:
pDG268neo_Beta-neoR
pDG268neo_GP35-neoR
pSB1C3_Beta-neoR
pSB1C3_GP35-neoR
Missing: figure tekst: Shows the general concepts of the two plasmids pDG268neo_recombinase and pSB1C3_recombinase. Both exists in two versions, one with each of the recombinase proteins CDSs (Beta and GP35). Those also have different RBSs since they are optimized for the CDS. Upstream of neoR is a promoter and RBS and downstream of neoR is a terminator, but sequences and positions of these features are not known.
For pDG268neo_recobinase a DNA sequence containing following features
Missing: make a table on following
● Promoter: PliaG from BBa_K823002 was used.
● RBSs were optimized for the specific CDS using the salis lab RBS calculator (Missing https://www.denovodna.com/software/)
● CDS for recombination protein beta or GP35
● Terminator: we use rho-independent Part:BBa_B0014
Sequence was ordered from IDT as two gblocks (for each recombinase) with overlapping regions, thus they can be assembled with Gibsom assembly.
pDG268neo was linearized in a PCR with primers, so that the native lacZ was omitted. This linearized plasmid was purified and used in a Gibsom assembly reaction with two cognate gblocks. This is shown on the figure missing.
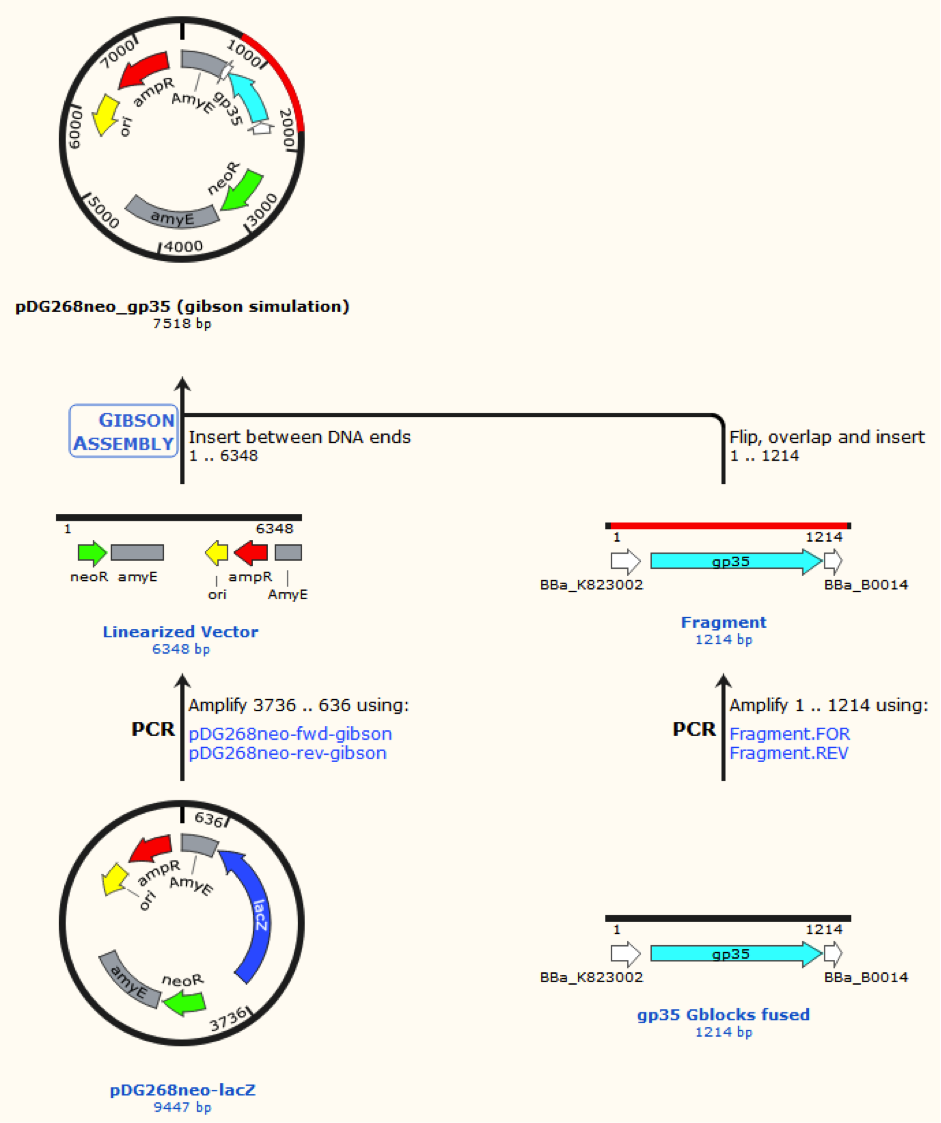
Missing figure text: Gibsom assembly of the pDG268neo_GP35, the gblocks was fused to the “gp35 Gblocks fused” in the same reaction. The Gibsom assembly of pDG268neo_beta was similar.
This resulted in two different plasmids pDG268neo_beta and pDG268neo_gp35.
The two mutS KO plasmids was made of the following DNA fragments:
1. About 500 bp up- and downstream from mutS was amplified, using primers with tails.
2. Recombinase and neoR expression cassettes was amplified from the pDG268neo_recombinase
3. Linearized pSB1C3 was used as template.
These fragments was assembled into two different plasmids pSB1C3_mutS::beta-neoR and pSB1C3_mutS::gp35-neoR using Gibsom assembly.
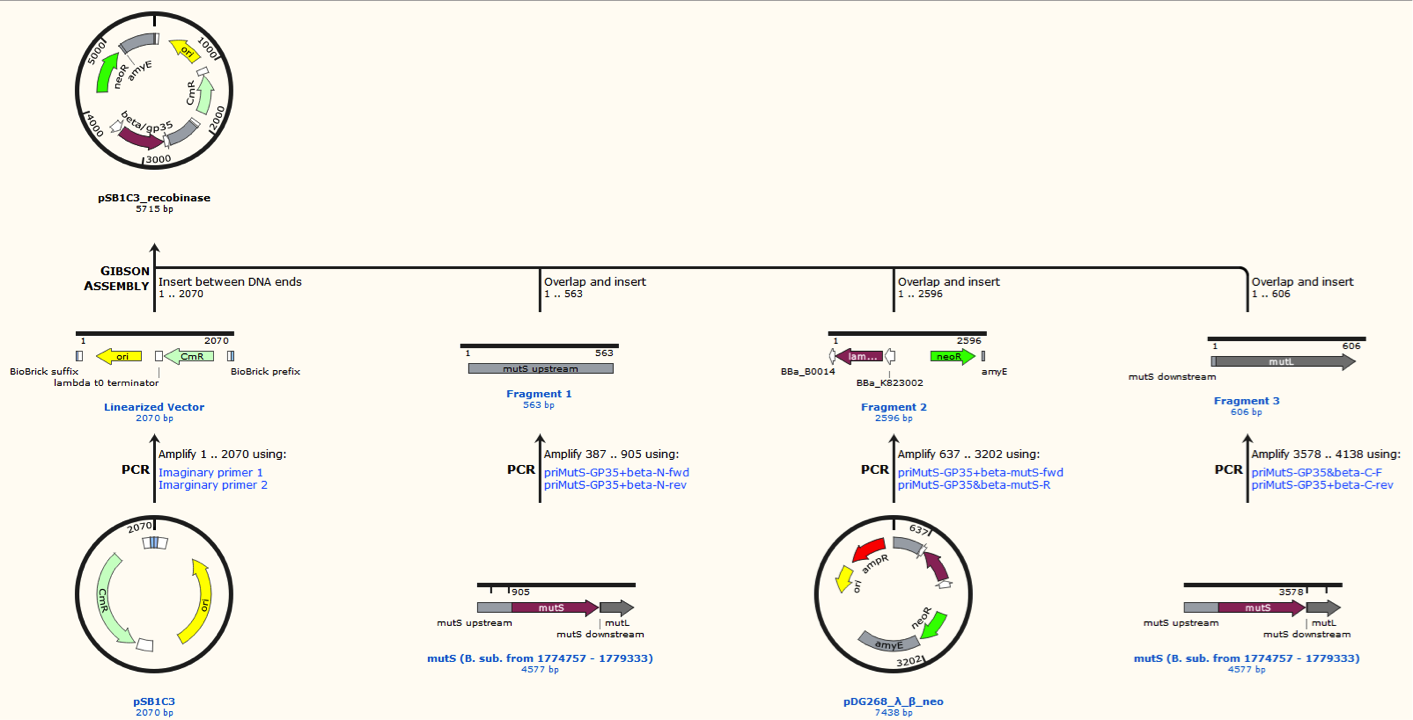
Missing figure text: Gibsom assembly of the pSB1C3_beta. The feature called “mutL” corresponding mutS downstream. The Gibsom assembly of pSB1C3_GP35 was similar.
All plasmids were verified by restriction enzyme digestion.
We prepared naturel competent B. subtilis 168 to be transformed. The plasmids were linearized with restriction enzymes. Then transformed into naturally competent B. subtilis 168, transformants were selected on 5ɣ neo. Transformants were verified with colony PCRs.
Conclusion
optimization of MAGE in B. subtilis
The MAGE method was optimized for Bacillus varying amount, length or amount.
Proof of concept of MAGE in B. subtilis with streptomycin
A single protein substitution in the ribosomal S12 protein in B. subtilis results in resistance to the antibiotic streptomycin. The required change is a lysine to arginine substitution at position 56 if the protein. The S12 subunit is coded by the rpsL gene (DOI 10.1099/mic.0.034595-01030 034595 MISSING). The strains were successfully transformed with oligos that incorporate the necessary mutation to create resistance. The mutS::beta showed to be the best mutant for inserting the oligos.
Method
Electrocompetent cells were prepared in accordance with the electrocompetent protocol (MISSING make link). Electropration was carried out using the electroporation protocol. The colonies from the LB plates were colonies picked unto an 500y streptomycin plate.
Results
|
|
total colonies |
transformants |
frequency |
|
ΔmutS::beta-neoR |
52 |
7 |
0.13 |
|
ΔmutS::GP35-neoR |
100 |
1 |
0.01 |
Table MISSING. mutS mutants transformation frequency for streptomycin resistance.
In the table shown above we can see that the OGRE method works. It seems that the Beta protein is better at inserting the mutation than the GP35 protein.
Protocol for multiple OGRE cycles in Bacillus subtilis 168
overview
We hypothesized that if the OGRE protocol was repeated multiple times the amount of transformants would rise. This was tested by running four cycles of the OGRE protocol. The progress could be followed by plating a dilution of the sample on streptomycin plates after every round and calculating the start value of the culture from the OD600 measurements. It was necessary to colony pick colonies onto streptomycin plates this gave usable results. The data is still not significant but it seems that more cycles does increase the amount of transformants.
Materials
The protocol specially med for this predure was followed ( MISSING see procol for multeple OGRE). This protocol takes aproximetly 6 hours for every cycle. Four cycles was run
Results
We had problems with finding the optimal dilutions. This cause the lack of data for some of the samples, new cells were replated from the glycerol stock of the differed OGRE cycles, but same problem was encountered again. There was a high variation in the amounts of transformants on the plates. We recommend to use the OD calculator ( MISSING make link) for making an approximation of the correct dilution factor.
 Figure MISSING. The frequency for the insertion of the streptomycin phenotype can be seen for the samples.
Figure MISSING. The frequency for the insertion of the streptomycin phenotype can be seen for the samples.
data can be seen here
It seems that mutS::beta is better than the wild type, mutS::GP35 and amyE::GP35. We cannot conclude that more OGRE cycles gives a higher yield than a single cycle, because the variation in the data is so great. The experiment will need to be run with a better method of testing if the insertion has been incorporated into the genome.
Achievements
Other part of mage
Optimal amount of oligo for OGRE
To optimize the OGRE protocol we tested different amount of oligo to find the optimal amount for transformation. The aim was to find the optimal amount of oligo for an OGRE cycle. Our experiment showed an unexpectedly high transformation efficiency of up to 50% efficiency. It seems unlikely that the transformation would be that efficient, when the efficiency in E. coli is 3.6% (Missing: Ref.). Also later experiments did not show the same high efficiency (missing link to mm experiment). The systematic errors, derived by the selection method, are thought to exceed the random errors. Consequently, more data will not make better results. The optimal approach will be to rethink the method measuring the efficiency.
Optimal amount of oligo for OGRE
· Designed a oligo for testing the OGRE method (should be in other section)
· Showed that 5uM is the optimal amount of oligo used
· Showed that exciding 5uM will decrease the efficency
Background
Here is the overview!!!!!!!! Victor!
Methods
Results
Methods
Different amounts of oligo were tested following the "OGRE in Bacillus subtilis 168" protocol. The oligo we used was B_Sub_Mods0007.1mutationrpsL(make to link to primers MISSING) introducing streptomycin resistance. The amount of oligo was varied between 0.05 - 6.25uM.
Results
| oligo amount uM | colonies picked | colonies growing on str | transformantion frequency | somthing else MISSING |
| 0.05 | 200 | 70 | 0.35 | |
| 1.25 | 300 | 54 | 0.18 | |
| 2.5 | 200 | 53 | 0.27 | |
| 5 | 200 | 98 | 0.49 | |
| 6.25 | 300 | 56 | 0.19 |
Table MISSING shows the transformation frequency of all colonies for an amount of oligo used.
The table above shows the transformation frequency. The transformation frequency varies from 18-50% this seems to be a too high number when comparing to earlier results and the standard transformation frequency for MAGE in E. coli. The data seems to suggest that the optimal oligo amount for transformation is 5 uM. This correlates with the optimal amount for E coli [1].
Selection of the colonies was difficult because of possible background from spontaneous mutants that became visible after 2 days and the risk of adding too much cell mass onto the plate when colony picking, this could be mistaken as a growing colony.
The results will need to be validated preferably using another screening method, since false positives seems to be an issue when using an antibiotic selection marker. It could be suggested to use a GFP with a inserted stop codon in the genome. The OGRE protocol could then be optimized for making knockins, this could be screened by using flow cytometry.
Finding frequency of mismatch insertion mutations
We tried to measure how the number of mismatches (missing: MM) effects the transformation efficiency.Oligos with one to six MM was examined. If successfully integrated, all the oligos would result in a streptomycin resistant strain.The results showed a very low efficiency, not following the trends implied in the proof of concept experiment or in the experiment doing multiple OGRE cycles (missing: link to proof of concept experiment. Missing: should refer to multiple cycle experiment, when it has not been mentioned above?).
Methods
Protocol for OGRE in Bacillus subtilis 168 (MISSING link) was followed using the six oligos with 1-6MM individually. Other parameters were kept constant (Missing: link to protocol and labnotes). Single colonies were taken from appropriate dilutions and colony picked onto 500ɣ strep. plates. Number re-streaked colonies that was growing on strep was counted. The transformation efficiency was calculated as the ratio between re-streaked colonies growing on LB and on 500ɣ strep.
Results
Three transformant were visible on the strep. plates with cells that had received the two mismatch oligoes, all other plates had no transformants after two days incubation.
The colony picked plates had a much higher rate of transformants. This is thought to be partly caused by a bias in the screening of the colony picked plates. (missing:). Unexpectedly the transformation frequency is higher for oligoes with more than three mismatches. Also the low transformation frequency is not in accordance with the previous results that showed a transformant rate of up to 100.
picture missing MISSING figure of colomb plot of frequency
It is possible that the cells were not electro competent. This experiment need to be redone to define if the experimental setup is incorrect or if the cells are not electro competent.
Optimizing oligo length
The length of the oliges was seen as an important contributor to the effectiveness of the transformation. We wanted to quantify the effect of the oligo length by using different oligoes with one mismatch that gave the cell streptomycin resistance. An extremely low transformation efficacy was observed. This was not in accordance with the previous results for streptomycin resistance.
Materials
The protocol for changing streptomycin ( MISSING insert link ) was used for oligo's with one mismatch but with varying lengths from 50-100nt were also tested. Because of low transformation efficiency colony picking was performed on the LB dilutions.
Results
Two transformants could be seen on the streptomycin plate plated with cells transformed with 60nt long oligo. All other streptomycin plates had no transformants. This low transformation rate contradicts the experiments done previously inducing streptomycin resistance. The colony picking showed better results than plating directly on 500y streptomycin (see figure below).
MISSING figure of colomb plot of frequency
The data is not sufficient to make accurate conclusions. The MISSING Figure shown above indicates that there is a negative correlation between length and transformation efficiency.
It is possible that the cells were not electro competent. This experiment need to be redone to define if the experimental setup is incorrect or if the cells are not electrocompetent.
Discussion
Conclusion
Dilution Equation
OD calculator
Overview
The Imperial iGEM 2008 team has made an equation for calculating CFU from OD600. We tried to validate their equation by using our own data. Unfortunately the variation in our results was too high to validate their equation. Based on their results we made a calculator that could compute the optimal dilution for plating to get a countable number of colonies.
Missing: Header2: Validating Imperial 2008s equation
Method
All the LB plate counts that we have done in our project was gathered and analyzed for this experiment. the data that was used can be seen here.
Results
The Imperial College team modulated following equation.
Missing: (Put op nice) Imperial equation: Y= 2*108 *X
Y = CFU/mL

Figure ? Our attempt to validate the Imperial College 2008 teams OD too CFU measurements. It is clear that the R2 value is not close optimal.
As can be seen from the plot ? shown below our data could not validate the equation completely. Though our data trends towards Imperials 2008s equation.
Missing: Header2: Dilution predictor
An equation for predicting a dilutions that will result in a countable number of CFUs was made from the Imperial College equation. The equation assume that 100µl is plated on a LB plate. The optimal amount of colonies is set to 150CFU on each plate.
Missing: Does the equations look nice?
YCFU=2*XOD*108
Yoptimal=150CFU/plate. This number can be varied to fit the user's preference.
d is the optimal dilution factor for getting 150CFU/plate. E.i. optimal dilution will be 10d.
\(Y_{optimal} = {{Y_{CFU} } \over {10^d* 10}}\)
\(Y_{optimal} = {{2*X_{OD}*10^8} \over {10^fd* 10}}\)
\(10^d= {{2*X_{OD}*10^8} \over {10*Y_{optimal}}}\)
\(d= log_{10}( {{2*X_{OD}*10^8} \over {10*Y_{optimal}}} ) , Y_{optimal}=150\)
\(d= log_{10}( {{2*X_{OD}*10^8} \over {10*150}} ) \)
\(d= log_{10}( {{1.33*X_{OD}*10^5} } ) \)
The formula has not been thoroughly test and the correlation between OD and CFU is low in for our data. Generally the formula overestimates dilution. Therefore we suggest that both 10d and 10d-1 are plated. A future solution to the problem could be to introduce a calibration constant to the right hand side of the equation. The constant can be fitted by rerunning the experiment with more samples.
References
- Carr, P. A., Wang, H. H., Sterling, B., Isaacs, F. J., Lajoie, M. J., Xu, G., … Jacobson, J. M. (2012). Enhanced multiplex genome engineering through co-operative oligonucleotide co-selection. Nucleic Acids Research, 40(17). doi:10.1093/nar/gks455
- Sun, Z., Deng, A., Hu, T., Wu, J., Sun, Q., Bai, H., … Wen, T. (2015). A high-efficiency recombineering system with PCR-based ssDNA in Bacillus subtilis mediated by the native phage recombinase GP35. Applied Microbiology and Biotechnology, 99(12), 5151–5162. doi:10.1007/s00253-015-6485-5
- Ginetti, F., Perego, M., Albertini, A. M., & Galizzi, A. (1996). Bacillus subtilis mutS mutL operon: Identification, nucleotide sequence and mutagenesis. Microbiology, 142(8), 2021–2029. doi:10.1099/13500872-142-8-2021

Department of Systems Biology
Søltofts Plads 221
2800 Kgs. Lyngby
Denmark
P: +45 45 25 25 25
M: dtu-igem-2015@googlegroups.com








