Difference between revisions of "Team:UESTC Software/Design.html"
| Line 65: | Line 65: | ||
<div class="safetyDiv design_div"> | <div class="safetyDiv design_div"> | ||
<div class="QandA"> | <div class="QandA"> | ||
| − | <h3 class="design_h3"> | + | <h3 class="design_h3">A reliable source of data</h3> |
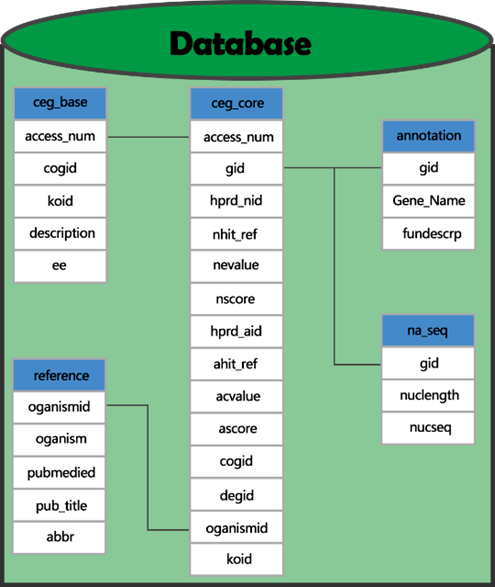
| − | <p> | + | <p>To deal with the data problem, we derived data of experimentally-determined essential genes from CEG database[1]. CEG(Cluster of Essential Gene) is a database which contains clusters orthologous essential genes. A notable feature of CEG is it deposits essential genes in orthologous groups rather than as single gene. There are seven tables in CEG database, and we used five of them selectively according to demand. Including ceg_core, which contains the information of each essential gene; ceg_base, which contains information of each gene cluster. And other three tables are reference, annotation and nucleic acid sequences.</p> |
| − | + | <p>We know upgrading is a key influence factor of the Product Life Cycle, and this is also our key consideration. The data of MCCAP will be constantly improving with updating of CEG, and reference species of it has increased from 16 to 29.</p> | |
| + | <img class="design_img" src="/wiki/images/8/84/Uestc_software_design.png" /> | ||
</div> | </div> | ||
<div class="QandA"> | <div class="QandA"> | ||
| − | <h3 class="design_h3"> | + | <h3 class="design_h3">Determining the minimal gene set</h3> |
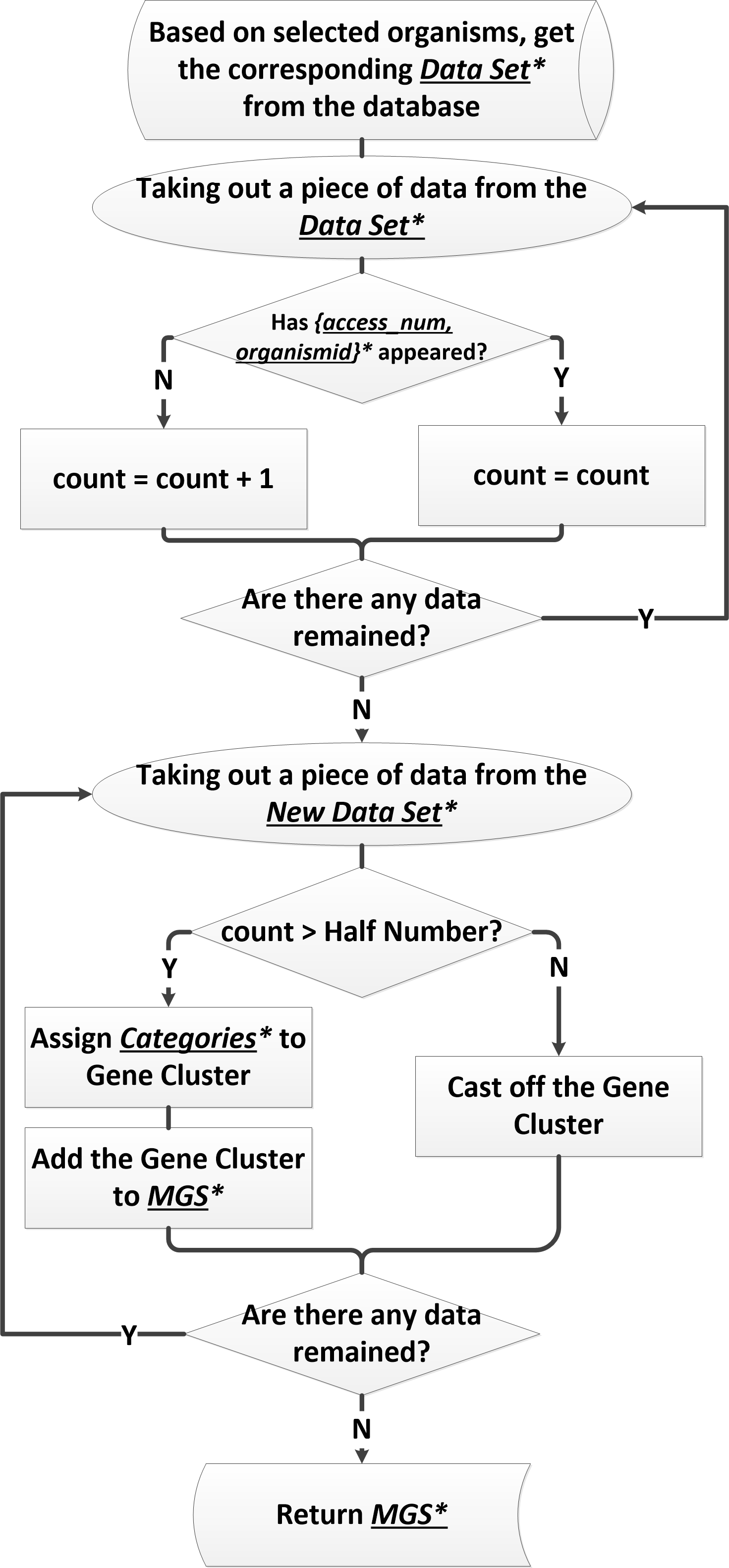
| − | <p>The | + | <p>The first and most important step of building a minimal artificial cell is determining the minimal gene set (minimal genome). Our method of solving this key task is the half retaining strategy. It is quite simple but effective. The implementer of the half-retaining strategy, MCCAP, achieves good results.</p> |
| − | < | + | <p><strong>While, what exactly is the half retaining strategy? </strong></p> |
| − | <p> | + | <p>Our half retaining strategy is basically one kind of Comparative Genomics Approach but it is further improved by starting with essential gene data determined by inactivation experiments. Firstly, we classify genes with the same function in different species into one category, named gene cluster. Secondly, we calculate the size of each gene cluster. Size means the number of species which contain the genes in this cluster. Finally, we put genes, whose size is no less than the half number (HN) together as the minimal gene set. For example, when there are 15 species, HN will be 8 according to our definition. So, if the size of a gene cluster is equal or more than 8, it will be added to the minimal gene set. In practice, we optimize the retaining ratio from the nominal half number to obtain more stable result.</p> |
| + | <p><strong>And what is difference between the half retaining strategy and traditional method?</strong></p> | ||
| + | <p>Contrast with the traditional comparative genomics method, it has two advantages as follow:</p> | ||
| + | <p>1. Traditional method is using comparative genomic approach to find ubiquitous genes from two completely sequenced small bacterial genomes. As we know in introduction, it will lose some genes when essential cellular function is performed by unrelated proteins that show no sequence similarities to each other in different organisms. However, half retaining strategy can get a stable result of minimal gene set, for it is based on plenty of reference species and an ideal retaining ratio.</p> | ||
| + | <p>2. Half retaining strategy uses experimentally-determined essential genes which are specific to organisms. Differ from traditional method, which is based on the whole genome, Half retaining strategy has lower false positive results.</p> | ||
| + | <p>MCCAP implements the half retaining strategy perfectly. Differ from traditional method, which provides a certain result of minimal gene set, MCCAP believe that minimal gene set is not unique, and it will be difference with different research fields. MCCAP provides a reference species selector interface to users, so that you can get a specific minimal gene set by your purposeful selection.</p> | ||
| + | <p>Most important information of minimal gene set to users is functions of genes. So in the results interface, MCCAP will give the function descriptions of gene firstly. MCCAP have also divided minimal gene set to four categories by their functions reference the standard of COG[2]. In fact, there are 25 function categories of COG. But, it is too complicated for user’s browsing experience. So MCCAP sum them up to four categories. These four categories are Transport and metabolism, Transcription and translation, General function prediction only and Others. </p> | ||
| + | <p>In addition to base information, MCCAP will also provide users with information related to KO(KEGG Orthology), EC number of enzymes, COG(Clusters of Orthologous Group) and so on. It is convenient to researchers to do further study.</p> | ||
| + | <img class="design_img" src="/wiki/images/3/30/Uestc_software_design_1.png"/> | ||
| + | <img class="" src="/wiki/images/a/ae/Uestc_software_design2_df.png"/> | ||
</div> | </div> | ||
<div class="QandA"> | <div class="QandA"> | ||
| − | <h3 class="design_h3"> | + | <h3 class="design_h3">Generating gene network diagram</h3> |
| − | + | <p>In addition to determining the minimal gene set, there is still a crucial consideration is to know gene metabolism and regulation in building a minimal artificial cell. In order to solve this problem, We combine KEGG Pathway and KEGG Module with the minimal gene set to generate gene network diagram. KEGG Pathway is a collection of manually drawn pathway maps representing our knowledge on the molecular interaction and reaction networks. KEGG Module is a collection of manually defined functional units[3]. </p> | |
| + | <p>Due to metabolism is the foundation by which organisms sustain their lives, we divided both pathway and module into two kinds. One is begin with Meta which means gene network diagram generated by only those genes related to metabolism. Another one is begin with Entire, which means gene network diagram generated by the whole minimal gene set.</p> | ||
| + | <p>For the functions of fetching information from KEGG, we reused the project of two previous iGEM teams. USTC_Software (<a href="https://2014.igem.org/Team:USTC-Software">https://2014.igem.org/Team:USTC-Software</a>) and Illinois-Tools (<a href="https://2009.igem.org/Team:Illinois-Tools">https://2009.igem.org/Team:Illinois-Tools</a>). </p> | ||
</div> | </div> | ||
<div class="QandA"> | <div class="QandA"> | ||
<h3 class="design_h3">Work flow of genetating the pathway and module</h3> | <h3 class="design_h3">Work flow of genetating the pathway and module</h3> | ||
| − | |||
<p>MACCP creates metabolism pathway by matching K value and using pathway and module database of KEGG. Because of the data that we use refers to the KEGG Orthology (KO) and Cluster of Orthologous Group (COG), not every gene cluster has K value. And we will abandon the gene cluster which doesn`t have K value because it could not create metabolism pathway.</p> | <p>MACCP creates metabolism pathway by matching K value and using pathway and module database of KEGG. Because of the data that we use refers to the KEGG Orthology (KO) and Cluster of Orthologous Group (COG), not every gene cluster has K value. And we will abandon the gene cluster which doesn`t have K value because it could not create metabolism pathway.</p> | ||
| + | <img class="design_img" src="/wiki/images/3/30/Uestc_software_design_2.png"/> | ||
| + | <img class="" src="/wiki/images/a/ae/Uestc_software_design1_df.png"/> | ||
</div> | </div> | ||
| + | <div class="QandA"> | ||
| + | <h3 class="design_h3">Processing gene to standard parts</h3> | ||
| + | |||
| + | <p>For researchers aiming to bring their design into reality, MCCAP provides informations related to standard parts and biobrick assembly standard. So that users can decide which certain gene to use and how to process this gene into a standard part. Currently, biobrick assembly standard supported by MCCAP contains RFC[10], RFC[12], RFC[21], RFC[23] and RFC[25].</p> | ||
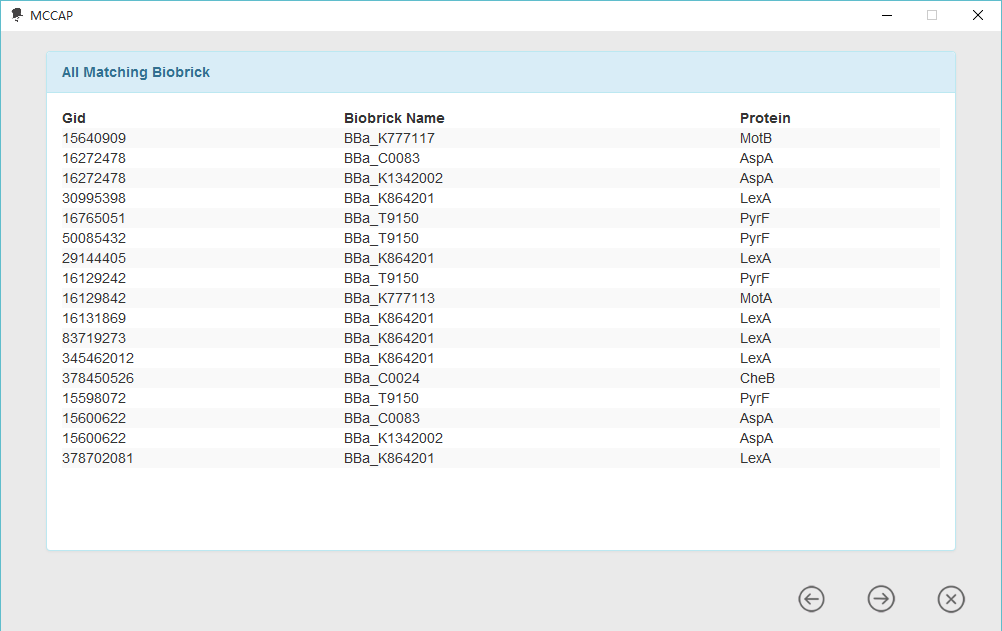
| + | <p>Besides, when considering the data, there may be some genes have been processed into standard parts. In order to avoid repeating the wheel, then we tried to find out them from protein coding sequences in registry of standard biological parts. if a gene from minimal gene set has already been processed into a biobrick, MCCAP will provide relevant information and we call it biobrick content. For this kind of gene is really seldom, we also list all of them in a table by considering the user’s convenience.</p> | ||
| + | <img class="design_img" src="/wiki/images/3/3c/Uestc_software_design4.png"/> | ||
| + | </div> | ||
| + | <div class="dou_ref"> | ||
| + | <p>Reference:<p> | ||
| + | <ul> | ||
| + | <li>[1] Ye Y N, Hua Z G, Jian H, et al. CEG: a database of essential gene clusters[J]. Bmc Genomics, 2013, 14(2):323-328.<li> | ||
| + | <li>[2] R L, Tatusov, M Y, Galperin, D A, Natale, et al. The COG database: a tool for genome-scale analysis of protein functions and evolution.[J]. Nucleic Acids Research, 2000, 28(1):: 33–36.<li> | ||
| + | <li>[3] Minoru, Kanehisa, Susumu, Goto, Yoko, Sato, et al. Data, information, knowledge and principle: back to metabolism in KEGG.[J]. Nucleic Acids Research, 2014, 42:D199-205.<li> | ||
| + | </div> | ||
</div> | </div> | ||
| − | + | ||
<footer> | <footer> | ||
<div class="footer_define"> | <div class="footer_define"> | ||
| Line 97: | Line 124: | ||
</div> | </div> | ||
</footer> | </footer> | ||
| + | </body> | ||
</html> | </html> | ||
Revision as of 15:46, 14 September 2015
<!doctype html>
Design
A reliable source of data
To deal with the data problem, we derived data of experimentally-determined essential genes from CEG database[1]. CEG(Cluster of Essential Gene) is a database which contains clusters orthologous essential genes. A notable feature of CEG is it deposits essential genes in orthologous groups rather than as single gene. There are seven tables in CEG database, and we used five of them selectively according to demand. Including ceg_core, which contains the information of each essential gene; ceg_base, which contains information of each gene cluster. And other three tables are reference, annotation and nucleic acid sequences.
We know upgrading is a key influence factor of the Product Life Cycle, and this is also our key consideration. The data of MCCAP will be constantly improving with updating of CEG, and reference species of it has increased from 16 to 29.
Determining the minimal gene set
The first and most important step of building a minimal artificial cell is determining the minimal gene set (minimal genome). Our method of solving this key task is the half retaining strategy. It is quite simple but effective. The implementer of the half-retaining strategy, MCCAP, achieves good results.
While, what exactly is the half retaining strategy?
Our half retaining strategy is basically one kind of Comparative Genomics Approach but it is further improved by starting with essential gene data determined by inactivation experiments. Firstly, we classify genes with the same function in different species into one category, named gene cluster. Secondly, we calculate the size of each gene cluster. Size means the number of species which contain the genes in this cluster. Finally, we put genes, whose size is no less than the half number (HN) together as the minimal gene set. For example, when there are 15 species, HN will be 8 according to our definition. So, if the size of a gene cluster is equal or more than 8, it will be added to the minimal gene set. In practice, we optimize the retaining ratio from the nominal half number to obtain more stable result.
And what is difference between the half retaining strategy and traditional method?
Contrast with the traditional comparative genomics method, it has two advantages as follow:
1. Traditional method is using comparative genomic approach to find ubiquitous genes from two completely sequenced small bacterial genomes. As we know in introduction, it will lose some genes when essential cellular function is performed by unrelated proteins that show no sequence similarities to each other in different organisms. However, half retaining strategy can get a stable result of minimal gene set, for it is based on plenty of reference species and an ideal retaining ratio.
2. Half retaining strategy uses experimentally-determined essential genes which are specific to organisms. Differ from traditional method, which is based on the whole genome, Half retaining strategy has lower false positive results.
MCCAP implements the half retaining strategy perfectly. Differ from traditional method, which provides a certain result of minimal gene set, MCCAP believe that minimal gene set is not unique, and it will be difference with different research fields. MCCAP provides a reference species selector interface to users, so that you can get a specific minimal gene set by your purposeful selection.
Most important information of minimal gene set to users is functions of genes. So in the results interface, MCCAP will give the function descriptions of gene firstly. MCCAP have also divided minimal gene set to four categories by their functions reference the standard of COG[2]. In fact, there are 25 function categories of COG. But, it is too complicated for user’s browsing experience. So MCCAP sum them up to four categories. These four categories are Transport and metabolism, Transcription and translation, General function prediction only and Others.
In addition to base information, MCCAP will also provide users with information related to KO(KEGG Orthology), EC number of enzymes, COG(Clusters of Orthologous Group) and so on. It is convenient to researchers to do further study.

Generating gene network diagram
In addition to determining the minimal gene set, there is still a crucial consideration is to know gene metabolism and regulation in building a minimal artificial cell. In order to solve this problem, We combine KEGG Pathway and KEGG Module with the minimal gene set to generate gene network diagram. KEGG Pathway is a collection of manually drawn pathway maps representing our knowledge on the molecular interaction and reaction networks. KEGG Module is a collection of manually defined functional units[3].
Due to metabolism is the foundation by which organisms sustain their lives, we divided both pathway and module into two kinds. One is begin with Meta which means gene network diagram generated by only those genes related to metabolism. Another one is begin with Entire, which means gene network diagram generated by the whole minimal gene set.
For the functions of fetching information from KEGG, we reused the project of two previous iGEM teams. USTC_Software (https://2014.igem.org/Team:USTC-Software) and Illinois-Tools (https://2009.igem.org/Team:Illinois-Tools).
Work flow of genetating the pathway and module
MACCP creates metabolism pathway by matching K value and using pathway and module database of KEGG. Because of the data that we use refers to the KEGG Orthology (KO) and Cluster of Orthologous Group (COG), not every gene cluster has K value. And we will abandon the gene cluster which doesn`t have K value because it could not create metabolism pathway.


Processing gene to standard parts
For researchers aiming to bring their design into reality, MCCAP provides informations related to standard parts and biobrick assembly standard. So that users can decide which certain gene to use and how to process this gene into a standard part. Currently, biobrick assembly standard supported by MCCAP contains RFC[10], RFC[12], RFC[21], RFC[23] and RFC[25].
Besides, when considering the data, there may be some genes have been processed into standard parts. In order to avoid repeating the wheel, then we tried to find out them from protein coding sequences in registry of standard biological parts. if a gene from minimal gene set has already been processed into a biobrick, MCCAP will provide relevant information and we call it biobrick content. For this kind of gene is really seldom, we also list all of them in a table by considering the user’s convenience.
Reference:
- [1] Ye Y N, Hua Z G, Jian H, et al. CEG: a database of essential gene clusters[J]. Bmc Genomics, 2013, 14(2):323-328.
- [2] R L, Tatusov, M Y, Galperin, D A, Natale, et al. The COG database: a tool for genome-scale analysis of protein functions and evolution.[J]. Nucleic Acids Research, 2000, 28(1):: 33–36.
- [3] Minoru, Kanehisa, Susumu, Goto, Yoko, Sato, et al. Data, information, knowledge and principle: back to metabolism in KEGG.[J]. Nucleic Acids Research, 2014, 42:D199-205.