Team:TU Eindhoven/Project/Design

Project - Design
 Home
Home Team
Team Project
Project Results
Results Modeling
Modeling Policy Practices
Policy Practices Collaborations
Collaborations Sponsors
Sponsors Safety
Safety


Dig Deeper
 Read how unnatural amino acids enable click chemistry
Read how unnatural amino acids enable click chemistry
Read how unnatural amino acids enable click chemistry
Next Chapter
 Explore how we tested our COMBs experimentally
Explore how we tested our COMBs experimentally
Explore how we tested our COMBs experimentally
Clickable Outer Membrane Biosensors - Design
We have developed Clickable Outer Membrane Biosensors as a universal membrane sensor platform for biosensors. Our dream is that any chassis may be functionalized with COMBs. This chassis should then be able to detect virtually any biomarker and translate detection of this biomarker into any desired response. To obtain such a system, it is of the utmost importance that the system is inherently modular. The modularity of our system is reached through rational choices for the recognition element, the scaffold and the signaling components for our COMBs.
The recognition elements - Aptamers
Molecular recognition is central to biosensing [1]. The moieties responsible for molecular recognition are better known as recognition elements. Recognition elements frequently used in biosensors are aptamers. These aptamers are small oligonucleotides which fold spontaneously into intricate three-dimensional structures. These structures fit perfectly into a wide range of disease markers, with affinities reaching into the low nanomolar range. Through modifications, these aptamers can be attached post-translationally to our proteins, resulting in a truly modular membrane sensor platform. Read more on aptamers:


Aptamers are folded oligo- nucleotides (orange) which perfectly fit into biomarkers (purple).
Image credit: Flanders' FOOD
Image credit: Flanders' FOOD
The scaffold - Outer Membrane Proteins
Outer membrane proteins form an essential component for Gram-negative bacteria, providing the bacteria with protection against a harsh environment [2]. More than six outer membrane protein families have been discovered, which all share a beta barrel secondary structure. Thanks to this structure, the proteins feature loops protruding from the bacterial outer membrane, and intracellular C-termini. This enables clicking of aptamers to the membrane proteins and fusing signaling components to the outer membrane proteins intracellularly, making outer membrane proteins the perfect scaffold for COMBs. Read more on outer membrane proteins:

OMPs can feature as a scaffold as aptamers can be clicked on the protruding loops & signaling components fused intracellularly.
The signaling components
The signaling components used for COMBs translate a close proximity into a response. Signaling components which do exactly this are split luciferases & fluorescent and bioluminescent proteins, through Resonance Energy Transfer. In the future, TEV Proteases may be used to translate a proximity into the release of the transcription factor, coupling COMBs recognition to the vast amount of cellular responses bacteria have available. Read more our signaling components:

COMBs signaling components translate a close proximity into a measurable signal. Here shown is BRET.
The Recognition Element – Aptamers 
Since the discovery of the nucleic acid structure, nucleic acids were long thought to have a single function – storage of the heredity information as genetic instructions. Our perception on the function of DNA and RNA changed radically, however, as it was discovered that small RNA molecules could fold into a three-dimensional structure, exposing a surface onto which other small molecules could perfectly fit. Soon after the discovery that nucleic acids could have interaction with other molecules, Craig Tuerk and Larry Gold described a procedure, called SELEX, to isolate high-affinity nucleic acid ligands for proteins through a Darwinian-like evolution process carried out in vitro [3].

 SELEX is an acronym for Systematic Evolution of Ligands by EXponential enrichment. The process is carried out in vitro and requires a very large oligonucleotide library (~1015 members), which can be generated by combinatorial DNA Synthesis. SELEX starts with incubation of the library with the target molecule. This incubation is followed by a washing step which ensures that only oligonucleotides with higher affinities are selected. Next, the oligonucleotides which are still present in the mixture after the washing step are recovered and amplified. During this amplification step, mutations are introduced into the oligonucleotides, yielding a new library. This library may again be incubated with the target molecule. Typically, these steps are repeated 8-15 times. In the last round of SELEX the oligonucleotides still present in the mixture are identified by sequencing.
SELEX is an acronym for Systematic Evolution of Ligands by EXponential enrichment. The process is carried out in vitro and requires a very large oligonucleotide library (~1015 members), which can be generated by combinatorial DNA Synthesis. SELEX starts with incubation of the library with the target molecule. This incubation is followed by a washing step which ensures that only oligonucleotides with higher affinities are selected. Next, the oligonucleotides which are still present in the mixture after the washing step are recovered and amplified. During this amplification step, mutations are introduced into the oligonucleotides, yielding a new library. This library may again be incubated with the target molecule. Typically, these steps are repeated 8-15 times. In the last round of SELEX the oligonucleotides still present in the mixture are identified by sequencing.
The Rise of Aptamers
25 Years after the discovery of Tuerk’s and Gold’s invention of SELEX, aptamers have become available for hundreds of ligands, including proteins, viruses, other small molecules and even whole cells.

Aptamer sequences have been published for over 500 ligands. These ligands include small molecules, viruses, proteins and whole cells. Among these are some very notable ligands, such as HIV, cancerous cells and ricin, the extremely toxic compound which plays a prominent role in the critically acclaimed Breaking Bad series. These figures were obtained through Aptamer Base, a collaboratively created knowledge base about aptamers and the experiments that produced them, curated and maintained by a team of aptamer researchers. For more information, visit Aptamer Base's website
A major limitation of aptamers in comparison with antibodies is their stability in vivo, where nucleic acids are rapidly degraded. Aptamers which have been used as therapeutic agents, for example, suffered from a half-life of 2 minutes [9]. This problem has, however, partially been overcome by using chemical modifications to the aptamers. An example of these modifications are Spiegelmers [8]. These oligonucleotides’ backbones contain L-Ribose instead of R-Ribose – “spiegel” means mirror. These mirrored aptamers are more stable in vivo, because they suffer less from degradation.
Aptamers in Biotechnology
Since aptamers can be easily modified chemically, they can be attached to numerous surfaces, enabling their way into a wide array of biosensors and drug delivery systems. Generally, the biosensors couple binding of the aptamer to a change in structure in the latter, which generates either a fluorescent or electrical signal. Aptamers have also found their way into drug delivery systems. A particular example of such a system is developed by Wu et al., featuring aptamers targeted at cancer cells with lipid tails. These aptamers could be used as building blocks of micelles, enabling efficient delivery of the micelles to cancerous cells [10] .
Aptamers in our Biosensor
Aptamers constitute the ideal sensor domain for our universal membrane sensor: they can be targeted at virtually any molecule, can easily be attached to the membrane using SPAAC chemistry and offer affinities similar to antibodies. Taking into account their remarkable stability and small size, they are unmatched by the traditional recognition elements of biotechnology, antibodies. An essential requirement for our system which limits the choice of aptamers is bifunctionality: the system relies on bringing two membrane proteins in close proximity by binding the ligand with two different moieties. Conceptually, this bifunctionality can be reached either through the use of dual aptamers or through the use of split aptamers.

Figure 1: Bifunctionality is required to bring the membrane proteins into close proximity.
Dual Aptamers
The first way in which bifunctionality can be reached is through the use of dual aptamers acting on different binding sites, called apitopes. A well-known example of a ligand for which dual aptamers are available is human thrombin. A drawback of the use of dual aptamers is that these are in general not available for small molecules and viruses, significantly reducing the available targets for the aptamers. The main target for dual aptamers are proteins.
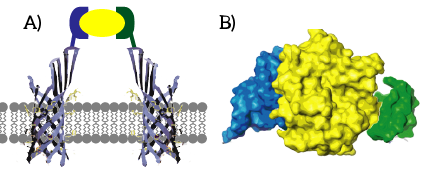
Split Aptamers
Alternatively, aptamers can be split into two parts. These parts can form heterodimers and thereby self-assemble into the three-dimensional structure required for binding the apitope. A crucial choice in engineering split aptamers is the choice of splitting site [11]. This choice is often not straightforward and the aptamer often suffers from a significantly decreased affinity. Obtaining split aptamers is, however, still in its infancy: of the approximately 100 small-molecule-binding aptamers, only six have been successfully engineered into split aptamers [11].

The Scaffold - OmpX
To realize conduction of a signal over the cell membrane, the aptamers have to be attached to a membrane protein. A procedure to attach oligonucleotides to membrane proteins has succesfully been tested by iGEM TU Eindhoven 2014. They presented this procedure as a part of their Click Coli project to the iGEM community. The procedure exploits the SPAAC reaction, which is one of the most well-described click chemistry reactions available (see Figure 4).

Figure 4: Strain-promoted Azide-Alkyne Cycloaddition (SPAAC) is a well known click chemistry reaction.
The reaction features two molecules, one functionalized with an azide group (A) and one functionalized
with a cyclooctyne group (B). SPAAC is very efficient and selective, no further reagents have to
be added to the reaction method and the reaction is completely bio-orthogonal. This image was adapted
from last year’s iGEM team’s wiki.
CPX & OmpX
CPX is a membrane display protein developed by Rice et al. as an alternative to traditional display methodologies, such as yeast display and phage display [12]. The protein itself is derived from the naturally occuring Outer membrane protein X (OmpX) through circular permutation (see Figure 5). Circular permutation was a necessary step to obtain the termini on the exterior of the bacterial cells.

Figure 5: CPX (right) is a membrane protein derived from OmpX (left) through circular permutation.
OmpX and CPX feature signal sequences (yellow) which ensure that the membrane protein is localized
to E.coli’s outer membrane. These signal sequences are cleaved from the peptides after localization. In
CPX, an amber stop codon was introduced to introduce a unnatural azide-functionalized amino acid.
Figure 6: The azide-functionalized amino acid enables clicking any DBCO-functionalized molecule to be attached to the cell membrane through SPAAC click chemistry. iGEM TU Eindhoven 2014 called this azide-functionalized CPX COMPx.


Figure 7: For our signaling proteins, we revert to the basis: OmpX. We functionalize OmpX by introducing the amber stop codon into its loops (red). The signaling domains are fused to OmpX C-terminally, since the N-terminus is occupied by the signaling sequence (yellow). The signaling sequence is cleaved from OmpX after localization.
The Loops
The amber stop codon will be incorporated in the protruding loops of OmpX. We have chosen to introduce the mutations into the loops since they are easily accessible and are not a part of the beta-barrel of OmpX [12]. Hence, we believe that mutations in the loops will not disturb the secondary structure of OmpX (see Figure 8B).

Figure 8: A) The OmpX protein structure has been elucidated through NMR and X-ray crystallography. B) The square residues are important for the secondary structure of OmpX. To keep the structure intact, we introduce an amber stop codon in one of the protruding loops. Figure 8B is adapted from [13].
The to be substituted residues in the loops were chosen in such a way that the clicked aptamer would protrude from OmpX. Moreover, residues were selected such that the signaling protein's structure closely resembles OmpX natural’s structure. The serine residue in loop 2 was chosen since it was the only available residue with side-groups pointing outward from OmpX. The tyrosine residue in loop 3 was chosen both because tyrosine closely resembles the unnatural azide-functionalized amino acid (see Figure 9) and because the tyrosine points outward from OmpX.

Figure 9: The unnatural azide-functionalized amino acid closely resembles tyrosine.
The Signaling Components
Quite paradoxically, the number of signaling proteins pales in comparison with the vast amount of cellular responses. This signaling paradox can be explained by the fact that cells employ amazingly complex signaling pathways, which feature signal amplification, signal integration and cross communication [14]. In addition to the vast amount of effects signaling pathways can have, these pathways operate over a staggering range of time scales; some signaling pathways act within less than a second, whereas others affect cell behavior only after hours or days.
An essential aspect of a universal membrane sensor is that it allows for these wide ranges of time scales and responses. We believe that the modularity of our membrane sensor and the complexity of intracellular cell signaling allows for a wide range of responses. To enable the sensors to act upon a wide range of time scales we consider multiple intracellular signaling components. These components all respond to a decrease in mutual distance of the membrane proteins as a result of ligand binding.
Fast Signaling Components - Exploiting Bioluminiscence
Two well-known principles to translate a close proximity into a measurable signal are Resonance Energy Transfer and the use of split luciferases. Even though these principles are very different, both response elements emit light, yielding a measurable signal. After ligand binding, these signaling components yield a virtually immediate response.

Figure 10: A) Resonance Energy Transfer and B) split luciferases both translate close proximity into a measurable signal in the form of light.
Resonance Energy Transfer is a physical process which can take place between fluorophores light emission when they are in close proximity (1-10nm) [15]. An electron which has been transferred to its ‘excited state’ falls back to its ‘ground state’. The energy which is released by the electron falling back to its ground state is normally released in the form of light. In the case of Resonance Energy Transfer, however, the energy of the electron falling back to its ground state is coupled to a transition of an electron in the RET Acceptor from its ground state to the excited state (see Figure 11).

Figure 11: Simplified energy-level diagram of RET. A) Normally, an excited electron falls back to its ground state under the emission of light (a radiative transition). B) In the case of Resonance Electron Transfer, an excited electron in the donor falls back to its ground state. This transition is coupled to the excitation of an electron in the acceptor. This excited electron falls back normally under the emission of light.
The efficiency of Resonance Energy Transfer is known to be dependent on a few factors, most importantly the relative orientation of the chromophores as well as the mutual distance of the chromophores [15]. A decreased mutual distance between the donor and acceptor increases the efficiency very significantly. This feature of Resonance Energy Transfer is exploited in the design of our membrane sensor. As a result of the reduced distance between the donor and acceptor, Resonance Energy Transfer is thus more efficient. Relatively more light will be emitted by the acceptor when the ligand is bound. The resulting signal is thus, per definition, ratiometric. Intensity of the light emitted by the donor will decrease and intensity of the light emitted by the acceptor will increase as a consequence of ligand binding (see Figure 12).

Figure 12: A) Efficient energy transfer is not possible since the donor and fluorophore are to remote. B) Efficient energy transfer is possible due to the proximity of the donor and acceptor. As a result, light intensity of the donor decreases whereas light intensity of the acceptor increases.
Split Luciferases
Luciferases are naturally occuring proteins which convert a substrate to a product and light. Many organisms feature luciferases to produce light, such as the sea spansy, fireflies and shrimps. These luciferases have been isolated from these organisms and can be succesfully used in vitro to produce light. Some luciferases have also been succesfully split into two complementary parts. These complementary parts can self-assemble into the functional luciferase, restoring its functionality to emit light by converting the substrate. As such, these split luciferases have become a major tool to study protein-protein interactions: these interactions are coupled to a close proximity of the proteins, allowing the parts to assemble into the functional luciferase and emit light (see Figure 13). We use these split luciferases in addition to BRET as a rapid signaling component.

Figure 13: A) The split parts of the luciferase are too remote to assemble into the functional luciferase. B) Upon ligand binding, the two luciferases can self-assemble into the functional luciferase. restoring its capability to produce light from the substrate, yielding a measurable signal.
[1] Chambers J.P., Arulanandam B.P., Matta L.L., Weis A. and Valdes J.J., “Biosensor recognition elements”, Curr. Issues Mol. Biol., vol. 10, no. 1, pp. 1–12, 2008.
[2] Koebnik R., Locher K.P. and Van Gelder P., “Structure and function of bacterial outer membrane proteins: barrels in a nutshell”, Mol. Microbiol., vol. 37, no. 2, pp. 239–53, Jul. 2000.
[3] Tuerk C. and Gold L., “Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase”, Science, vol. 249, no. 4968, pp. 505–10, Aug. 1990.
[4] Musumeci D. and Montesarchio D., “Polyvalent nucleic acid aptamers and modulation of their activity: A focus on the thrombin binding aptamer”, Pharmacol. Ther., vol. 136, no. 2, pp. 202–15, Jul. 2012.
[5] Cibiel A., Pestourie C. and Ducongé F., “In vivo uses of aptamers selected against cell surface biomarkers for therapy and molecular imaging”, Biochimie, vol. 94, no. 7, pp. 1595–1606, Feb. 2012.
[6] Lauridsen L.H. and Veedu R.N., “Nucleic acid aptamers against biotoxins: a new paradigm toward the treatment and diagnostic approach”, Nucleic Acid Ther., vol. 22, no. 6, pp. 371–79, Oct. 2012.
[7] Rhouati A., Yang C., Hayat A. and Marty J.L., “Aptamers: A promosing tool for ochratoxin a detection in food analysis”, Toxins (Basel)., vol. 5, no. 11, pp. 1988–2008, Nov. 2013.
[8] Radom F., Jurek P.M., Mazurek M.P., Otlewski J. and Jeleń F., “Aptamers: Molecules of great potential”, Biotechnol. Adv., vol. 31, no. 8, pp. 1260–74, Apr. 2013.
[9] Hicke B.J. et al., “Tumor Targeting by an Aptamer”, J. Nucl. Med., vol. 47, no. 4, pp. 668–78, Apr. 2006.
[10] Wu Y., Sefah K., Liu H., Wang R. and Tan W., “DNA aptamer-micelle as an efficient detection/delivery vehicle toward cancer cells”, Proc. Natl. Acad. Sci. U. S. A., vol. 107, no. 1, pp. 5–10, Nov. 2009.
[11] Kent A.D., Spiropulos N.G. and Heemstra J.M., “General approach for engineering small-molecule-binding DNA split aptamers”, Anal. Chem., vol. 85, no. 20, pp. 9916–23, Oct. 2013.
[12] Rice J.J., Schohn A., Bessette P.H., Boulware K.T. and Daugherty P.S., “Bacterial display using circularly permuted outer membrane protein OmpX yields high affinity peptide ligands”, Protein Sci., vol. 15, no. 4, pp. 825–36, Apr. 2006.
[13] Vogt J. and Schulz G.E., “The structure of the outer membrane protein OmpX from Escherichia coli reveals possible mechanisms of virulence”, Structure, vol. 7, no. 10, pp. 1301–9, Oct. 1999.
[14] Alberts, Johnson, Lewis, Raff, Roberts, Molecular Biology of The Cell. Pearson, 2005.
[15] Medintz I. and Hildebrandt N., Eds., FRET - Förster Resonance Energy Transfer. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA, 2013.
[2] Koebnik R., Locher K.P. and Van Gelder P., “Structure and function of bacterial outer membrane proteins: barrels in a nutshell”, Mol. Microbiol., vol. 37, no. 2, pp. 239–53, Jul. 2000.
[3] Tuerk C. and Gold L., “Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase”, Science, vol. 249, no. 4968, pp. 505–10, Aug. 1990.
[4] Musumeci D. and Montesarchio D., “Polyvalent nucleic acid aptamers and modulation of their activity: A focus on the thrombin binding aptamer”, Pharmacol. Ther., vol. 136, no. 2, pp. 202–15, Jul. 2012.
[5] Cibiel A., Pestourie C. and Ducongé F., “In vivo uses of aptamers selected against cell surface biomarkers for therapy and molecular imaging”, Biochimie, vol. 94, no. 7, pp. 1595–1606, Feb. 2012.
[6] Lauridsen L.H. and Veedu R.N., “Nucleic acid aptamers against biotoxins: a new paradigm toward the treatment and diagnostic approach”, Nucleic Acid Ther., vol. 22, no. 6, pp. 371–79, Oct. 2012.
[7] Rhouati A., Yang C., Hayat A. and Marty J.L., “Aptamers: A promosing tool for ochratoxin a detection in food analysis”, Toxins (Basel)., vol. 5, no. 11, pp. 1988–2008, Nov. 2013.
[8] Radom F., Jurek P.M., Mazurek M.P., Otlewski J. and Jeleń F., “Aptamers: Molecules of great potential”, Biotechnol. Adv., vol. 31, no. 8, pp. 1260–74, Apr. 2013.
[9] Hicke B.J. et al., “Tumor Targeting by an Aptamer”, J. Nucl. Med., vol. 47, no. 4, pp. 668–78, Apr. 2006.
[10] Wu Y., Sefah K., Liu H., Wang R. and Tan W., “DNA aptamer-micelle as an efficient detection/delivery vehicle toward cancer cells”, Proc. Natl. Acad. Sci. U. S. A., vol. 107, no. 1, pp. 5–10, Nov. 2009.
[11] Kent A.D., Spiropulos N.G. and Heemstra J.M., “General approach for engineering small-molecule-binding DNA split aptamers”, Anal. Chem., vol. 85, no. 20, pp. 9916–23, Oct. 2013.
[12] Rice J.J., Schohn A., Bessette P.H., Boulware K.T. and Daugherty P.S., “Bacterial display using circularly permuted outer membrane protein OmpX yields high affinity peptide ligands”, Protein Sci., vol. 15, no. 4, pp. 825–36, Apr. 2006.
[13] Vogt J. and Schulz G.E., “The structure of the outer membrane protein OmpX from Escherichia coli reveals possible mechanisms of virulence”, Structure, vol. 7, no. 10, pp. 1301–9, Oct. 1999.
[14] Alberts, Johnson, Lewis, Raff, Roberts, Molecular Biology of The Cell. Pearson, 2005.
[15] Medintz I. and Hildebrandt N., Eds., FRET - Förster Resonance Energy Transfer. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA, 2013.




