Difference between revisions of "Team:Oxford/Blog/The-how-tos-of-construct-design"
Weikongquee (Talk | contribs) |
Weikongquee (Talk | contribs) |
||
| (2 intermediate revisions by the same user not shown) | |||
| Line 13: | Line 13: | ||
</style> | </style> | ||
| − | <p style="font-size: | + | <p style="font-size:9px"; class="caption">Clicking on the image above redirects you to our original Wordpress blog post</p> |
<br> | <br> | ||
| Line 23: | Line 23: | ||
} | } | ||
| − | + | h1 { | |
| + | text-align: center; | ||
| + | } | ||
| + | |||
| + | p.welcome { | ||
| + | text-align: center; | ||
| + | |||
| + | } | ||
| + | |||
| + | |||
| + | p.text { | ||
| + | text-align: justify; | ||
| + | } | ||
| + | |||
| + | span.blue { | ||
| + | color: #0000FF; | ||
| + | } | ||
| + | |||
| + | span.pink { | ||
| + | color: #ff69b4; | ||
| + | } | ||
| + | |||
| + | IMG.displayed { | ||
| + | display: block; | ||
| + | margin-left: auto; | ||
| + | margin-right: auto } | ||
| + | |||
| + | table, td { | ||
| + | border: 1px solid black; | ||
| + | } | ||
| + | |||
| + | #footer { | ||
| + | float:bottom; | ||
| + | width: 97.9%; | ||
| + | text-align: center; | ||
| + | padding-left: 20px; | ||
| + | border-top: 2px solid #002147; | ||
| + | border-bottom: 2px solid #002147; | ||
| + | background-color: #002147; | ||
| + | } | ||
| + | |||
| + | |||
| + | span.white { | ||
| + | color: #fff; | ||
| + | text-decoration:none; | ||
| + | font-weight: 200; | ||
| + | font-size: 9px; | ||
| + | } | ||
| − | |||
p.date { | p.date { | ||
| Line 84: | Line 130: | ||
<p>This is the stage we are at with our project. Most of the Easter vacation was spent finalising our project and the last few weeks have involved sorting our DNA sequences and playing around with SnapGene. Currently, we’re making sure that our constructs comply with IDT DNA synthesis rules and double checking that everything is in the right place and right order to put into plasmids when the DNA arrives. After adding the final touches to our constructs we should be ready to order by the end of next week.</p> | <p>This is the stage we are at with our project. Most of the Easter vacation was spent finalising our project and the last few weeks have involved sorting our DNA sequences and playing around with SnapGene. Currently, we’re making sure that our constructs comply with IDT DNA synthesis rules and double checking that everything is in the right place and right order to put into plasmids when the DNA arrives. After adding the final touches to our constructs we should be ready to order by the end of next week.</p> | ||
| + | |||
| + | </div> | ||
| + | |||
| + | |||
| + | |||
| + | </div> | ||
| + | |||
| + | </html> | ||
| + | |||
| + | {{OxfFooter}} | ||
Latest revision as of 17:25, 9 June 2015

Clicking on the image above redirects you to our original Wordpress blog post
THE HOW-TOS OF CONSTRUCT DESIGN
Written by Raphaella Hull on May 8 2015
One of the main aims of genetic engineering is to get bacteria to make proteins that they cannot normally synthesise. For bacteria to be able to do this, we must take the DNA that codes for these new proteins and transfer it into bacteria, so that they can understand it and make proteins from it. One of the ways that DNA can be moved into bacteria is by using a plasmid.
A plasmid is a circular piece of DNA. The DNA that codes for the new proteins can be added into a plasmid, which can then be put into bacteria (through a process called transfection) and, providing the plasmid contains the all necessary extra features for gene expression, the bacteria will be able to make the brand new proteins. For example, human insulin is made using bacteria that have been genetically-engineered in this way. It used to be that the only way to obtain insulin was purifying it from the pancreas of cows and pigs that had been slaughtered for food. These days, we can put the gene for human insulin into a plasmid, put this plasmid into bacteria and have the bacteria make insulin for us.

{kind=link}
The DNA sequences that you want to add to a plasmid can come from a variety of sources. In the simplest case, the sequence you are looking for is already a biobrick, such as the protein DNase. For this component of our project, we used the iGEM parts registry to locate the DNA sequence. Many of the parts we need to use for our project are already biobricks. This includes the promoter regions, which are regions of DNA that initiate the expression of a particular gene (in effect an on/off switch), and the ribosome binding sites that facilitate the initiation of protein synthesis. If the sequence you need is not a biobrick, you can find it on online databases such as GenBank.
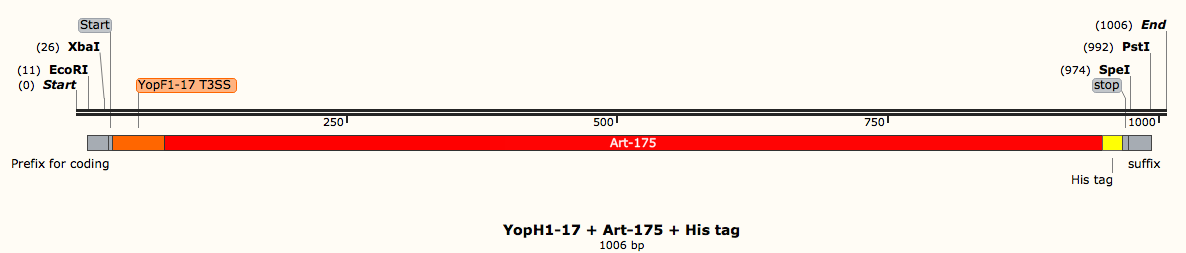
In other cases, you may have the amino acid sequence of a protein you want to express, but not the DNA sequence that you need to insert into the plasmid. For us, this was the case with the protein Art-175. The paper from which we sourced the sequence of Art-175 only gave the amino acid sequence. We used IDT codon optimization to get the DNA sequence. This software gives the DNA sequence when you enter the amino acid. This programme also optimizes the DNA sequence for expression in E. coli (or whichever bacteria species you are wanting to express your proteins of interest in). This is an important step, for which I will now describe the biochemistry.
Amino acids are the building blocks of proteins. There are 20 amino acids and each is coded for by three DNA bases, which are called codons. As there are 4 different nucleotides (A,T,C,G), there are 64 different possible codons. With only 20 amino acids, this means that some amino acids are coded for by more than one codon. For example, AGA, AGG, AGT and ACG all code for the amino acid serine. The relative frequency of codon use varies widely depending on the organism and organelle. For example, AGA is the most commonly used codon for serine in E. coli. When taking the amino acid sequence of the protein you want to express in E. coli, the codon optimization software gives the DNA sequence using the codons that are most commonly used by E. coli. This is important to avoid very slow or incorrect protein synthesis.

When you have the codon optimised DNA sequences, restriction enzymes are then used to cut and stick the sequences and plasmids. These enzymes cut at specific sequences of DNA called restriction sites and allow us to stick our pieces of DNA together. iGEM specifies certain restriction sites at the start and end of DNA sequences so that any sequences can be cut and stuck together using only a few restriction enzymes. If the restriction enzyme sites were present within any of the DNA sequences (and not only within the prefixes and suffixes) then the restriction enzymes would cut within and mix up all the sequences, rendering them useless. Before using restriction enzymes, it is therefore crucial to check all of the DNA sequences for any disallowed restriction enzyme sites.
For example, the DNA sequence where the EcoRI restriction enzyme cuts is GAATTC. This sequence is present in the DNA sequence for the protein Dispersin B, meaning that EcoRI would cut within the coding sequence, leading to a shortened amino acid sequence and a nonfunctional protein. With this in mind, it is necessary to change the codon to alter the restriction site whilst at the same time ensuring that the same amino acid is coded for (a different amino acid means an incorrect protein). GAA codes for glutamic acid but so goes GAG; thus by changing the third nucleotide from A to G, the final polypeptide sequence remains unchanged (glutamic acid is still coded for) but there is no longer an EcoRI restriction site within the DspB coding sequence.

Once you have all the DNA sequences for everything you need, it’s then time to assemble your constructs in a software like SnapGene. Together, the coding DNA sequences and the control elements make the constructs that need designing. For example, a part of our project looks at different ways of secreting anti-biofilm agents out of E. coli. If we’re considering artilysin (one of the proteins we want to secrete), the construct needs to contain the DNA sequence for artilysin as well a secretion signal (a tag that directs the protein to the secretion system), a histidine tag (enables us to purify the protein at a later stage) and start and stop sequences (enable the bacteria to know where to start reading the DNA and were to stop). We put all of these parts together in the right order so that the bacteria can understand what to make and how to make it. These constructs have to be made for each individual component of the project. The DNA order can be made after ensuring that the constructs are suitable for DNA synthesis by a company such as IDT.
This is the stage we are at with our project. Most of the Easter vacation was spent finalising our project and the last few weeks have involved sorting our DNA sequences and playing around with SnapGene. Currently, we’re making sure that our constructs comply with IDT DNA synthesis rules and double checking that everything is in the right place and right order to put into plasmids when the DNA arrives. After adding the final touches to our constructs we should be ready to order by the end of next week.
Contact us:
E-mail: oxfordigem@bioch.ox.ac.uk | Mailing Address: Department of Biochemistry, University of Oxford, South Parks Road, Oxford OX1 3QU, United Kingdom

![]()
![]()