Difference between revisions of "Team:KU Leuven/Modeling/Top"
(added discretized system + lightbox) |
|||
| Line 15: | Line 15: | ||
src="http://cdn.mathjax.org/mathjax/latest/MathJax.js?config=TeX-AMS-MML_SVG.js"> | src="http://cdn.mathjax.org/mathjax/latest/MathJax.js?config=TeX-AMS-MML_SVG.js"> | ||
</script> | </script> | ||
| + | |||
| + | <script src="http://yourjavascript.com/11518521655/lightbox.js" | ||
| + | type="text/javascript"></script> | ||
<script> | <script> | ||
Revision as of 14:04, 15 September 2015

1-D continuous model
The video above shows how the proposed method for pattern formation works. Two cell types A and B are interacting. Type
A cells produce a repellent called leucine which causes the cells of type B to move away. At the same time type A cells
also produce AHL, which is required by the cells of type B to move. Initially, colonies of the two cell types are placed
at the center of the dish. As molecule production within the type A cells kicks in, the repellent and AHL concentrations
start to increase. This triggers the type B cells to move away from the center. Movement will continue until the concentration of AHL is insufficient for the type B cells to move further.
Our Keller-Segel type model
$$\frac{\partial A}{\partial t} = D_a \bigtriangledown^2 A + \gamma A(1 - \frac{A}{k_{p}}),$$ $$\frac{\partial B}{\partial t} = D_b \bigtriangledown^2 B + \bigtriangledown (P(B,H,R) \bigtriangledown R)+ \gamma B(1 - \frac{B}{k_{p}}), $$ $$ \frac{\partial R}{\partial t} = D_r \bigtriangledown^2 R + k_r A - k_{lossH} R $$ $$\frac{\partial H}{\partial t} = D_h \bigtriangledown^2 H + k_h A - k_{lossR} H . $$With:
$$ P(B,H,R) = \frac{-B K_{c} H}{R}. $$
The model has been derived while looking at [1] and [2] .
The terms that appear can be grouped into four categories. Every equation has a diffusion term given by
$D_x \bigtriangledown^2 X$, diffusion smoothes peaks by spreading them out in space. The two equations related to cell
densities contain logistic growth terms of the form $\gamma X(1-\frac{X}{k_x})$, which model the cell growth during
simulation time. Finally the second equation describing the moving cells comes with a variable coefficient Poisson term
$\bigtriangledown (P \bigtriangledown X)$ which describes
To generate the video file above the system above has been discretized using a finite volume approach in conjunction,
with an explicit Euler scheme, For finite volume methods to work we rewrite our equations as conservation laws. Then each grid
point is assigned the area around it such that flux of cells or molecules leaving one cell enters another one. From
discretizing the integrated conservation law the following expression is obtained in one dimension:
Discretized Keller-Segel type model
$$ A^{n+1}_j = A^n_j + \triangle t \cdot (D_a/(\triangle x)^2 \cdot ( A^n_{j-1} + A^n_{j+1} - 2 \cdot A^n_j)) ... $$ $$ + \gamma \cdot A^n_j \cdot (1 - A^n_j / kp)) $$ $$B^{n+1}_j = B^n_j +\triangle t \cdot (1/ (\triangle x)^2 \cdot (D_b\cdot (B^n_{j-1} + B^n_{j+1} - 2B^n_j)\dots $$ $$ -(P^n_{j+\frac{1}{2}} \cdot (R^n_{j+1} - R^n_j) - P^n_{j-\frac{1}{2}} \cdot (R^n_j - R^n_{j-1}))) \dots $$ $$ + \gamma \cdot B^n_j \cdot (1 - B^n_j / kp)) $$ $$ R^{n+1}_j = R^n_j + \triangle t \cdot( D_r \cdot (R^n_{j+1} + R^n_{j-1} - 2 R^n_j) /(\triangle x^2) \dots $$ $$ + kr \cdot A^n_j - k_{lossR} \cdot R^n_j) $$ $$ H^{n+1}_j = H^n_j + \triangle t \cdot ( D_h \cdot (H^n_{j+1} + H^n_{j-1} - 2 H^n_j) / (\triangle x)^2 \dots $$ $$ + k_h \cdot A^n_j - k_{lossH} \cdot H^n_j ) $$The for the equations given above the left hand side values at the next time step depend exclusively on data of the prefious time step as illustrated in the figure below:
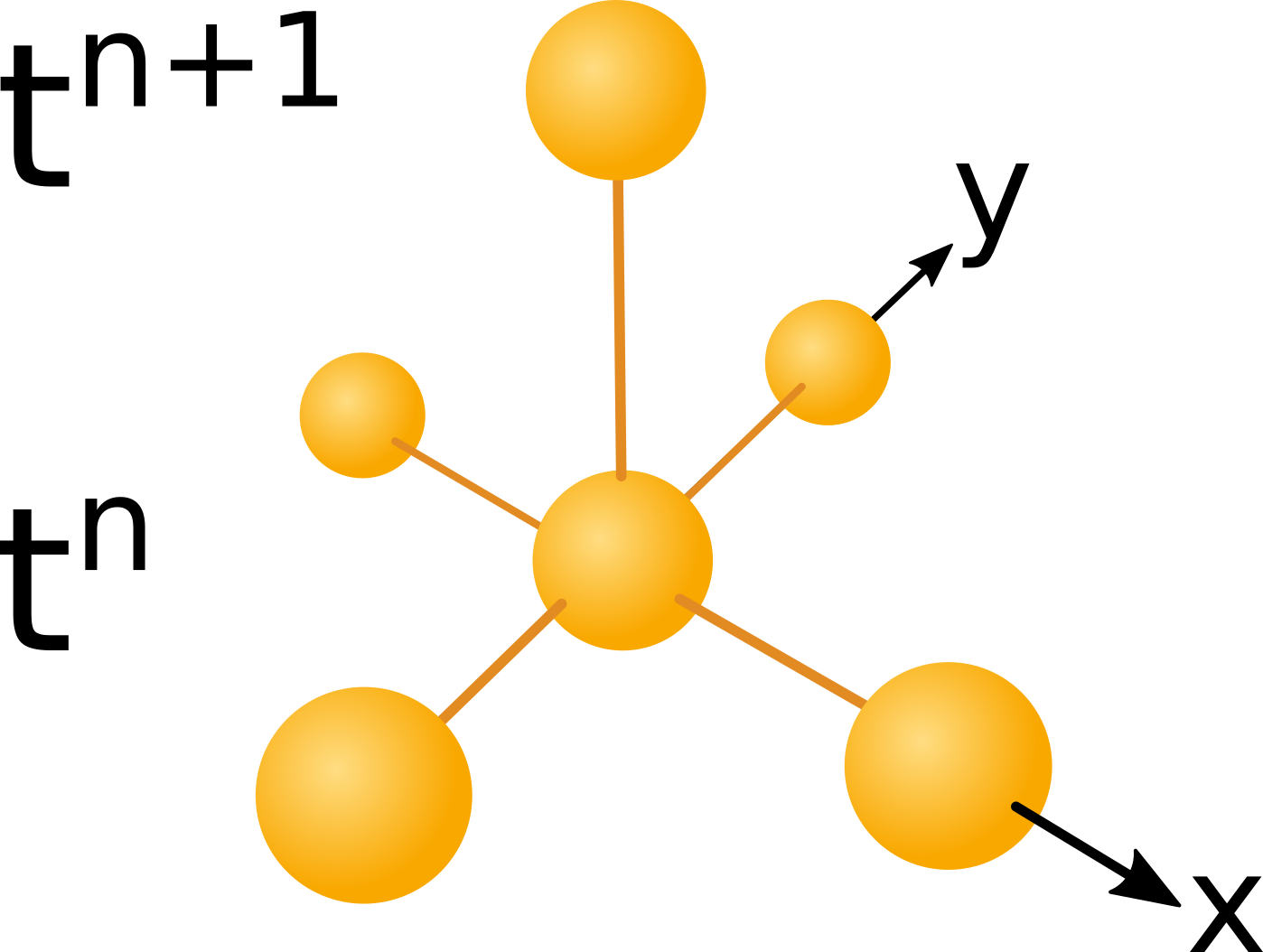
Figure 2
computational molecule. Click to enlarge
The image above shows the dependency of data in time and space. The computational molecule used in this case uses only data of
the previous time level $t_n$ to compute data at the next time level $t_{n+1}$. A scheme with a space time dependency like the
one shown above is called an explicit scheme.
Zero flux and periodic boundary conditions have been implemented. The boundaries are the edges of the domain on which the
equation system is solved. Here the domain ranges from zero to eight centimetres, which is the diameter of a Petri dish.
With zero flux boundaries
the first derivative is set to zero at the boundaries, which means that neither bacteria nor chemicals are allowed to pass
trough the boundary. Periodic boundaries connect pairs of boundaries to each other, which means that cells leaving at the top
of the boundary appear at the bottom,
cells leaving at the left boundary reappear at the right and so on. In the continuous context these boundary conditions have
been implemented to allow comparisons with the hybrid model, where these boundaries are also used.
Finally simulation has been done using the parameters given in the table below:
| Parameter | Value | Unit | Source | Comment |
|---|---|---|---|---|
| $D_a$ | $0.072 \cdot 10^{-3}$ | $cm^2/h$ | following [1] | |
| $D_b$ | $2.376 \cdot 10^{-3}$ | $cm^2/h$ | following [1] | |
| $D_r$ | $26.46 \cdot 10^{-3}$ | $cm^2/h$ | as found in [6] | $298.2 K$ |
| $D_h$ | $50 \cdot 10^{-3}$ | $cm^2/h$ | from [3] | |
| $K_{c}$ | $8.5 \cdot 10^{-3}$ | $cm^2 \cdot cl/h$ | estimated | |
| $\gamma$ | $10^{-5}$ | $h^{-1}$ | from [1] | |
| $k_p$ | $1.0 \cdot 10^2$ | $cl^{-1}$ | from [1] | |
| $k_h$ | $17.9 \cdot 10^{-4}$ | $fmol/h$ | computed from [4] and [8] | |
| $k_r$ | $5.4199\cdot 10^{-4}$ | $fmol/h$ | computed from [7] and [8] | |
| $k_{lossH}$ | $ln(2)/48$ | $h^{-1}$ | from [5] | $ ph = 7$ |
| $k_{lossR}$ | $ln(2)/80$ | $h^{-1}$ | estimated |
2-D continuous model
Using the equation system as described above, the model may also be simulated in two dimensions. Once more a finite volume approach has been taken in connection with an explicit Euler scheme. All parameters have been kept constant with the one exception of the chemotactic sensitivity $K_c$. Which has been increased to $Kc = 1.5 * 10^{-1}$
References
| [1] | D. E. Woodward, R. Tyson, M. R. Myerscough, J. D. Murray, E. O. Budrene, and H. C. Berg. Spatio-temporal patterns generated by Salmonella typhimurium. Biophysical journal, 68(5):2181-2189, May 1995. [ DOI | http ] |
| [2] | Benjamin Franz and Radek Erban. Hybrid modelling of individual movement and collective behaviour. Lecture Notes in Mathematics, 2071:129-157, 2013. [ http ] |
| [3] | Monica E Ortiz and Drew Endy. Supplement to- 1754-1611-6-16-s1.pdf, 2012. [ .pdf ] |
| [4] | A. B. Goryachev, D. J. Toh, and T. Lee. Systems analysis of a quorum sensing network: Design constraints imposed by the functional requirements, network topology and kinetic constants. In BioSystems, volume 83, pages 178-187, 2006. [ DOI ] |
| [5] | A. L. Schaefer, B. L. Hanzelka, M. R. Parsek, and E. P. Greenberg. Detection, purification, and structural elucidation of the acylhomoserine lactone inducer of Vibrio fischeri luminescence and other related molecules. Bioluminescence and Chemiluminescence, Pt C, 305:288-301, 2000. |
| [6] | Tatsuya Umecky, Tomoyuki Kuga, and Toshitaka Funazukuri. Infinite Dilution Binary Diffusion Coefficients of Several α-Amino Acids in Water over a Temperature Range from (293.2 to 333.2) K with the Taylor Dispersion Technique. Journal of Chemical & Engineering Data, 51(5):1705-1710, September 2006. [ DOI ] |
| [7] | Xuejing Yu, Xingguo Wang, and Paul C. Engel. The specificity and kinetic mechanism of branched-chain amino acid aminotransferase from Escherichia coli studied with a new improved coupled assay procedure and the enzyme's potential for biocatalysis. FEBS Journal, 281(1):391-400, January 2014. [ DOI ] |
| [8] | Yasushi Ishihama, Thorsten Schmidt, Juri Rappsilber, Matthias Mann, F Ulrich Hartl, Michael J Kerner, and Dmitrij Frishman. Protein abundance profiling of the Escherichia coli cytosol. BMC genomics, 9:102, 2008. [ DOI ] |




Contact
Address: Celestijnenlaan 200G room 00.08 - 3001 Heverlee
Telephone n°: +32(0)16 32 73 19
Mail: igem@chem.kuleuven.be



