Team:Waterloo/Lab/sgRNA
Simple sgRNA Exchange
Introduction
sgRNA is a chimera of tracrRNA and crRNA, which were the original two-part biomolecule that allowed Cas9 to find and identify its target dsDNA .
The purpose of fusing together the tracrRNA and crRNA was to simplify the CRISPR-Cas9 system for scientists to use more easily. Aligning with what the University of Waterloo is best known for, the lab team continued this innovation by spending part of its summer re-engineering sgRNA so that the twenty base pair target region could be removed and replaced by a new twenty base pair target region with basic classical cloning techniques. This challenge was tackled by first investigating the architecture of the sgRNA scaffold region, followed by a study of how different point mutations to the sgRNA scaffold region would affect its secondary structure. Both tasks were completed by a thorough scientific literature review that lead to the final, characterized design shown further below.

Design
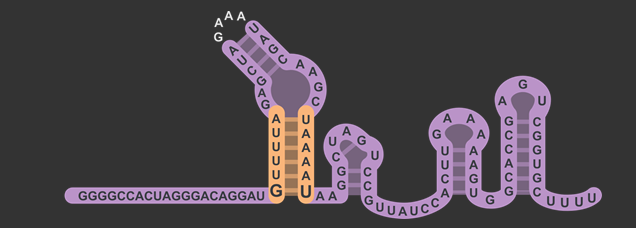
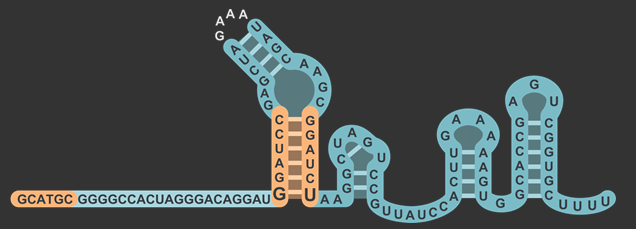
In total, there were three different modifications made to both the target and scaffold regions of the sgRNA. SphI was added so that it would flank the target region on the left side, and BamHI was added so that it would flank the target region on the right side. BamHI’s base pairs are integrated into the scaffold region, and based on how the secondary structure is folded, the downstream sequence 5’-GGATC-3’ was modified to properly accommodate the new BamHI restriction enzyme site (RE site).The original five base pairs were 5’-TAAAA-3’. It is also worth noting that thymine was not changed, which will be explained in the following section about the rationale for this design.
Theoretical Validation
Although there were only three modifications made to sgRNA’s sequence, many uncertainties had to be elucidated before sending the modified sgRNA sequence for synthesizing. This section will address the following questions whose answers will validate the design of the sgRNA modification. Briner et al.’s article on sgRNA’s secondary structure and how it guides Cas9 activity played an integral role in answering all these questions .

The target sequence is located on the bottom right of this figure with a length of 20 base pairs. Immediately following this is the lower stem, also known as the first anti-repeat region, coloured green and made of six base pairs running up bonded to another six pairs downstream running down. As mentioned earlier, the lower stem contains a non-Watson-Crick base pair between the guanosine and uracil at the bottom of the lower stem. The yellow bulge follows after and links the second anti-repeat region, coloured blue, to the first (viz. the bulge attaches the lower stem to the upper stem). At the top of this component is the 5’-GAAA-3’ tetraloop that combines what was originally the tracrRNA and the crRNA. The downstream section of the lower stem is connected to the nexus, coloured pink, followed by the double hairpin components coloured in grey . Overall, the sgRNA without the U6 promoter and transcriptional terminator is a hundred base pairs.
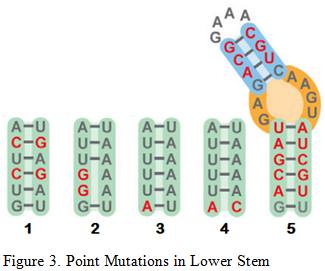
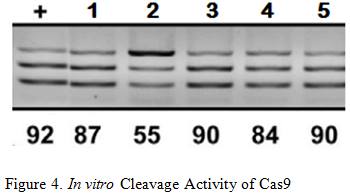
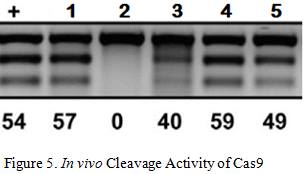
Briner et al. showed how the five mutations above affected Cas9’s cleavage rate in both in vitro and in vivo experiments . The in vitro experiments used Cas9 and transcribed sgRNA mixed together in a stabilizing buffer. The in vivo experiments consisted of the Cas9 and mutated sgRNA transfected into the HEK-293 cell line.
Mutation 1 converts A-T base pairs to G-C base pairs to test how stronger hydrogen bonding would affect the cleavage rate. Figure 3 shows that Mutation 1 allowed Cas9 to retain a strong cleavage rate both in vitro and in vivo. Since the BamHI sequence introduces new G-C base pairs into the lower stem, mutation 1 provides evidence that this modification should be safe.
Mutation 2 tests the effect of having non-matching base pairs in the lower stem, resulting in a significant drop in in vitro cleaving activity at 55%, as well in vivo cleavage activity at 0%. This information reinforces the need to ensure that the downstream section of the lower stem is as complementary as possible to the upstream section for cleaving activity to be retained.
Mutation 3 and 4tests the effects of mutating the base of the lower stem, yielding similar results that indicate Cas9 cleavage activity is retained. This information means that the last base pair of the lower stem can be freely mutated to other base pairs without significant loss in the cleavage rate, which is necessary for the design of the BamHI Complement.
Arguably the most remarkable and humorous observation was Mutation 5 where multiple base pairs in both the lower and upper stem were simultaneously mutated to demonstrate the tolerance of this structural component. Retaining an in vitro cleavage rate of 90% and in vivo cleavage rate of 49% strongly suggests that the three modifications made to the sgRNA will have minimal influence on Cas9’s cleavage activity since the only mutation made are substitutions and not indel mutations .
Why isn’t the BamHI Complement fully complementary to the upstream section of the lower stem?
The goal of the modification is to only swap out to target sequence by performing a double digest with SphI and BamHI. This poses two problems. The first is that the plasmid containing the sgRNA-Mod must not have SphI and BamHI sites anywhere else, otherwise more than one cut will be made. This problem was solved by choosing a plasmids for construction that did not have SphI or BamHI sites. The second problem is that having a lower stem where both sections are fully complimentary would introduce a second BamHI site making the target sequence swap a messy experiment. However, based on Mutation 3 and 4, the base of the lower stem can remain a non-Watson-Crick pairing which means that the downstream section of the lower stem does not have to be fully complementary to the BamHI cut site on the upstream section of the lower stem. This eliminates the second BamHI site and thus makes the swapping mechanism possible with the target sequence.
Why SphI and BamHI? Would other enzymes have worked too?
Indeed other enzymes may have worked, but there are important considerations that were made when choosing these two RE sites. The first consideration is whether the cut would introduce a blunt end or a sticky end. It was decided that blunt end RE sites should not be used in order to avoid the possibility of adding the target sequence backwards. A mistake like this would cause Cas9 to have off-target cleaving activity, and thus an overall poor experiment. Both SphI and BamHI produce sticky ends.
The second consideration is the type of sticky end the REs would produce. It was decided that having the same overhangs flanking the target sequence could lead to unintended circularization of the plasmid, ultimately preventing the new target sequence from becoming part of the sgRNA. SphI produces a 5’ overhang, and BamHI produces a 3’ overhang. This difference in overhangs is anticipated to improve ligation efficiency because of the inability of the overhangs to re-attach.
The third consideration is the ease-of-use and cost of the REs. The following table compares the price of the standard BioBrick REs compared to SphI and BamHI.
All prices were calculated using Fisher Scientific CAD prices by taking the cost of the largest bulk order possible and dividing it by the RUs provided by that bulk order. For example, EcoRI’s largest bulk order of 20’000 RUs costs $132.30. Dividing the cost by the RUs reveals that one single digest reaction with EcoRI will cost about a sixth of a cent. BamHI is cheaper than all the standard BioBrick REs, and SphI costs as much as the most expensive standard BioBrick RE, SpeI, at 12 cents per reaction. The protocols for using both SphI and BamHI in digests do not differ significantly from the BioBrick REs, assuming that the all mentioned REs are from the same bioproduct.
Combining the answers to these four questions, it can be confidently hypothesized that the three modifications made to the sgRNA will have minimal effect on Cas9 cleavage activity. In light of this better understanding of sgRNA’s architecture and the relevant point mutation effects, the following constructs were used to build the necessary plasmids for the experiments.
Experimental Constructs
- Dual Color Target
- sgRNA-Mod RFP
- sgRNA-Mod GFP
- sgRNA-Ctrl RFP
- sgRNA-Ctrl GFP
- dCas9
Wet Lab Validation
The experiments to validate the sgRNA-Mod design can be divided into two sets. Set A’s experiments are designed to characterize the sgRNA-Mod prototype, and Set B’s experiments are designed to show the feasibility of swapping out one target sequence with another one. Both sets are needed to have full confidence in sgRNA-Mod’s ability to minimize the need to synthesize multiple sgRNAs with different target sequences. The following does not include the steps made to create the constructs; it only contains the experiments that follow after making the constructs.
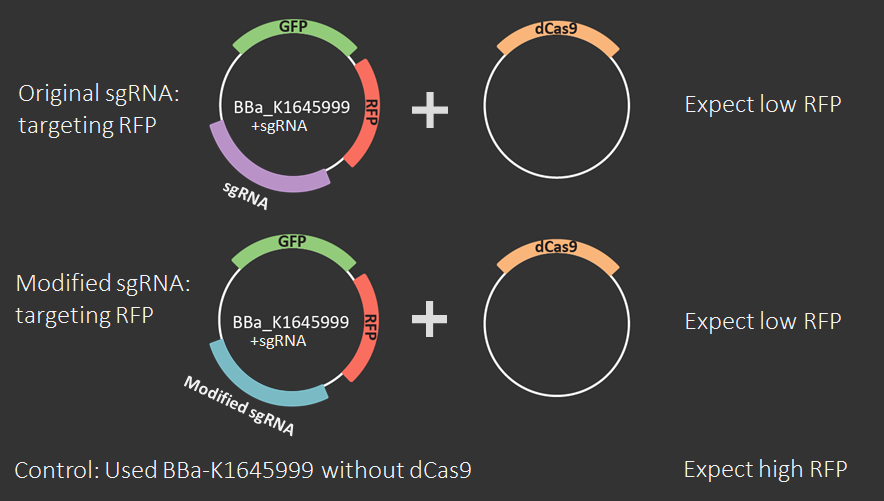
Set A
This set is the most important because it characterizes the sgRNA-Mod design using flow cytometry experiments. Set B is less important because the only difference between these two sets is that Set A becomes Set B through a simple SphI-BamHI double digest, ligation, transformation into DH5α, and miniprep.Part 1
- Co-transform sgRNA-Mod-RFP with dCas9 into BL21(DE3).
- Inoculate transformants into 5mL of SOB media containing antibiotic selection and induce with 5uL of IPTG. Incubate for 14hrs or more.
- Inoculate transformants into 5mL of SOB media containing antibiotic selection, but do not induce with IPTG. Incubate for same amount of time as step 2.
- Remove samples from incubator after the incubation period and run a flow experiment.
Part 2
- Co-transform sgRNA-Ctrl-RFP with dCas9 into BL21(DE3).
- Inoculate transformants into 5mL of SOB media containing antibiotic selection and induce with 5uL of IPTG. Incubate for 14hrs or more.
- Inoculate transformants into 5mL of SOB media containing antibiotic selection, but do not induce with IPTG. Incubate for same amount of time as step 2.
- Remove samples from incubator after the incubation period and run a flow experiment.
Part 3
- Transform sgRNA-Ctrl-RFP into BL21(DE3).
- Inoculate transformants into 5mL of SOB media containing antibiotic selection and induce with 5uL of IPTG. Incubate for 14hrs or more.
- Inoculate transformants into 5mL of SOB media containing antibiotic selection, but do not induce with IPTG. Incubate for same amount of time as step 2.
- Remove samples from incubator after the incubation period and run a flow experiment.
Set B
As mentioned earlier, Set A and Set B are the same except for the following changes.Part 1
- Co-transform sgRNA-Mod-GFP with dCas9 into BL21(DE3).
- Inoculate transformants into 5mL of SOB media containing antibiotic selection and induce with 5uL of IPTG. Incubate for 14hrs or more.
- Inoculate transformants into 5mL of SOB media containing antibiotic selection, but do not induce with IPTG. Incubate for same amount of time as step 2.
- Remove samples from incubator after the incubation period and run a flow experiment.
Part 2
- Co-transform sgRNA-Ctrl-GFP with dCas9 into BL21(DE3).
- Inoculate transformants into 5mL of SOB media containing antibiotic selection and induce with 5uL of IPTG. Incubate for 14hrs or more.
- Inoculate transformants into 5mL of SOB media containing antibiotic selection, but do not induce with IPTG. Incubate for same amount of time as step 2.
- Remove samples from incubator after the incubation period and run a flow experiment.
Part 3
- Transform sgRNA-Ctrl-GFP into BL21(DE3).
- Inoculate transformants into 5mL of SOB media containing antibiotic selection and induce with 5uL of IPTG. Incubate for 14hrs or more.
- Inoculate transformants into 5mL of SOB media containing antibiotic selection, but do not induce with IPTG. Incubate for same amount of time as step 2.
- Remove samples from incubator after the incubation period and run a flow experiment.
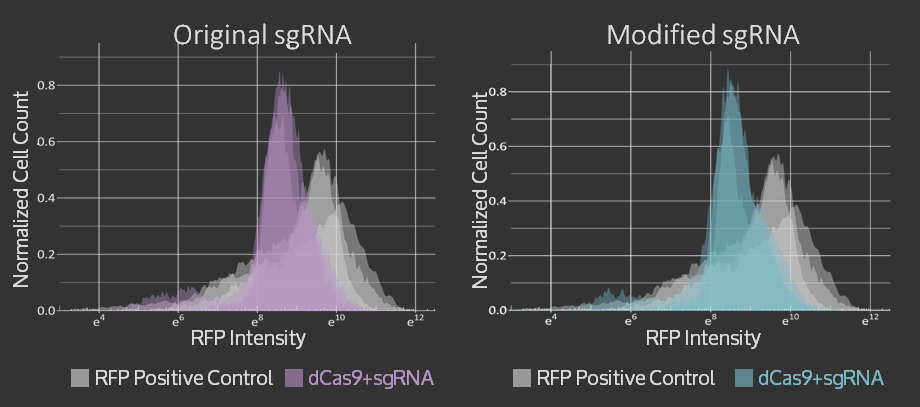
Characterization & Results
An amnis Imaging Flow Cytometry assay was used to obtain these results. Cell cultures with dCas9 + original sgRNA, dCas9 + modified sgRNA and no dCas9 were imaged and RFP fluorescence from 10000 cells was measured. In both cases that include dCas9, the mean RFP fluorescence intensity (plotted on the x-axis of the graphs below) is shifted to the left, i.e. to lower intensities, compared to the control. The triplicate measurements are technical replicates measured on different days.