Difference between revisions of "Team:Waterloo/Modeling/Cas9 Dynamics"
(add formation) |
|||
| Line 50: | Line 50: | ||
<h3>Probability of Double-Stranded Cuts made by CRISPR/Cas9</h3> | <h3>Probability of Double-Stranded Cuts made by CRISPR/Cas9</h3> | ||
| − | + | <p>The probability of a target being cut in a given time step was modelled as dependent on Cas9 concentration and sgRNA mismatches. The average time for a cut was also considered using the reported from 2-minute half-life Hemphill et al. <cite ref="Hemphill2015"></cite>.</p> | |
| − | + | ||
| − | + | <p>Since Cas9 binds to PAM sites according to an approximately first order <cite ref="Qi2013"></cite> three dimensional diffusion <cite ref="Sterberg2014"></cite> we expect increasing concentration of Cas9 protein to lead to a higher probability of cutting. Concentration was incorporated into our model using a multiple regression on data from Kusku et al.(2014) <cite ref="Kusku2014"></cite>, which related the proportion bound as a function of concentration and number of mismatches between the target and the sgRNA.</p> | |
| − | + | ||
| − | + | <p>The effect of mismatches in the sgRNA (important for the residual targeting after indels have been introduced by NHEJ) was further considered using the relationship found by Hsu et al. <cite ref="Hsu2013"></cite>. These effects were assumed to be independent, which likely overestimates mismatch effects and underestimated CRISPR/Cas9 efficacy.</p> | |
| − | <h3> | + | <h3>Error-Prone Repair by Non-Homologous End Joining</h3> |
| − | + | ||
| + | <p>Open breaks in the DNA were repaired according to an exponential decay, following the model of Reynolds et al. <cite ref="Reynolds2012"></cite>, which found that DSBs remained with a half-life of 8 minutes. Insertion and deletion sizes were chosen based on a distributions of indels observed by deep-sequencing of repaired targets (see data in <cite ref="Crispresso"></cite>). When there is a net insertion, we selected from a uniform distribution of [ACGT] to add new nucleotides</p>. | ||
| + | |||
| + | <h3>Large Deletions</h3> | ||
| + | <p>The probability of large deletions was estimated using a study on large deletions, which measured the percentage of large deletions observed at 3 days and 10 days at several targets <cite ref="Ousterout2015"></cite>. We chose not to account for the effect of the distance between targets for large deletions, though it could be incorporated in future studies <cite ref="Canver2014"></cite>.</p> | ||
| − | <p> | + | <p>We expect there to be multiple viral genomes in our plant defense example and it is possible that simultaneous cuts on different genomes could result two genomes being joined together. However, we chose to disregard this possibility and averaged results from multiple stochastic simulations could be averaged to get an overall picture of gene deactivation by Cas9.</p> |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</section> | </section> | ||
Revision as of 03:24, 19 September 2015
Modeling Genomic Effects of CRISPR/Cas9
CRISPR/Cas9 has been extensively studied for its applications in eukaryotic genome editing and gene expression control. Last year, the Waterloo iGEM team created an ODE model of dCas9 binding and control of gene expression. This year, however, the modelling team chose to investigate the effects of CRISPR/Cas9 on an genomic rather than molecular level. Specifically, we wanted to model the accumulation of mutations in a target genome and eventual deactivation of target genes after cutting by CRISPR/Cas9 and repair by Non-Homologous End Joining (NHEJ).

Model Formation
When bound to a single guide RNA (sgRNA), the S. pyogenes Cas9 nuclease diffuses through the cell in three dimensions, searching for the sequence 'NGG' in the target genome . When it finds an 'NGG', known as a PAM site, Cas9 binds and undergoes a conformational change that allows it to unwind the DNA helix and compare the sequence of its sgRNA with the DNA. If the sgRNA matches well, Cas9 cleaves the DNA, producing a double-stranded break (DSB) 3-4 bp upstream of the PAM site .
In the absence of a template, DSBs are repaired by Non-Homologous End Joining (NHEJ), which is an error-prone process that sometimes creates indels at the site of repair . This effect has recently been exploited to target double-stranded viruses such as HBV . Though there have been extensive efforts to characterize the factors that contribute to effective targeting and deactivation by CRISPR/Cas9 and NHEJ, they have not, to the best of our knowledge, been synthesized into a single model.
The aim of the model is thus to capture the cutting events initiated by Cas9 and predict the outcomes of these events. We model each genome as containing multiple domains of interest, such as promoters or ORFs, and track whether these domains have been deactivated by CRISPR/Cas9 activity. There may be more than one sgRNA target in each domain and many domains can be targeted at once.


If Cas9 successfully cuts at a target site, the double-stranded break may be resolved in three ways. The most common resolution is for NHEJ to successfully repair the DSB without creating any indels
However, NHEJ repair is error-prone and will often induce indels at the target site. Finally, since multiple sgRNA targets are considered, it is possible that large deletions will occur between two targets that are simultaneously cut.

At each timestep, the model considered the state (cut or uncut) and sequence of all targets and computes the probability of the following events at each target: CRISPR/Cas9 cutting, NHEJ repair or large deletion. The remainder of the model formation section discusses how we determined the probability of each event.
Probability of Double-Stranded Cuts made by CRISPR/Cas9
The probability of a target being cut in a given time step was modelled as dependent on Cas9 concentration and sgRNA mismatches. The average time for a cut was also considered using the reported from 2-minute half-life Hemphill et al. .
Since Cas9 binds to PAM sites according to an approximately first order three dimensional diffusion we expect increasing concentration of Cas9 protein to lead to a higher probability of cutting. Concentration was incorporated into our model using a multiple regression on data from Kusku et al.(2014) , which related the proportion bound as a function of concentration and number of mismatches between the target and the sgRNA.
The effect of mismatches in the sgRNA (important for the residual targeting after indels have been introduced by NHEJ) was further considered using the relationship found by Hsu et al. . These effects were assumed to be independent, which likely overestimates mismatch effects and underestimated CRISPR/Cas9 efficacy.
Error-Prone Repair by Non-Homologous End Joining
Open breaks in the DNA were repaired according to an exponential decay, following the model of Reynolds et al. , which found that DSBs remained with a half-life of 8 minutes. Insertion and deletion sizes were chosen based on a distributions of indels observed by deep-sequencing of repaired targets (see data in ). When there is a net insertion, we selected from a uniform distribution of [ACGT] to add new nucleotides
.Large Deletions
The probability of large deletions was estimated using a study on large deletions, which measured the percentage of large deletions observed at 3 days and 10 days at several targets . We chose not to account for the effect of the distance between targets for large deletions, though it could be incorporated in future studies .
We expect there to be multiple viral genomes in our plant defense example and it is possible that simultaneous cuts on different genomes could result two genomes being joined together. However, we chose to disregard this possibility and averaged results from multiple stochastic simulations could be averaged to get an overall picture of gene deactivation by Cas9.
Software Implementation
Genome Classes
The code uses three classes to model the genome. Genomes have domains which have targets. Targets handle probabilities, domains track functionality and genome modifies everything.
class Target():
is associated with a domain
class Domain():
has targets
is associated with a genome
class Genome():
has domains
Genome Simulation
The simulation calls these classes to check if events have occurred and the details of each event. At the end it compiles the data logs into CSVs, plots and visualizations.
for dt in time_steps:
call genome_classes to check if there was a cut, repair or large deletion
if event:
add to log
generate CSVs, plots and visualizations
To see all the code for the simulation, check out our GitHub Page
Results
Model Validation
Include notes on how the model matches reality/our expectations of reality in this section.
Simulate w/ targets that mismatch to different extents.
Effect of sgRNA Strength
Matt visualizations for different sgRNAs.
Graph of 3 different sgRNA designs of different strengths, show % functional
Importance of Large Deletions
Include notes on how the model matches reality/our expectations of reality in this section.
Effect of Cas9 Concentration
Include notes on how the model matches reality/our expectations of reality in this section.
Predicting CRISPR Plant Defense
This model was applied to the CRISPR Plant Defense aspect of our project, investigating whether the P6 protein of Cauliflower Mosaic Virus (CaMV) could be deactivated by frameshift mutations. The P6 protein was chosen as a focus of the investigation because it suppresses natural plant RNAi defenses and trans-activates translation of other CaMV proteins . Details on P6 and the CaMV genome can be found on CaMV Biology page.
The model was run with three targets in the P6 gene of the simulated CaMV genome described. The particular sgRNA target locations in P6 are those described as Design II on the related wet lab page. We tracked the percent of simulated genomes with functional P6 across 1000 runs fo the model, giving a general prediction of how long it will take before the P6 of a particular CaMV genome is rendered non-functional by our Plant Defense system.
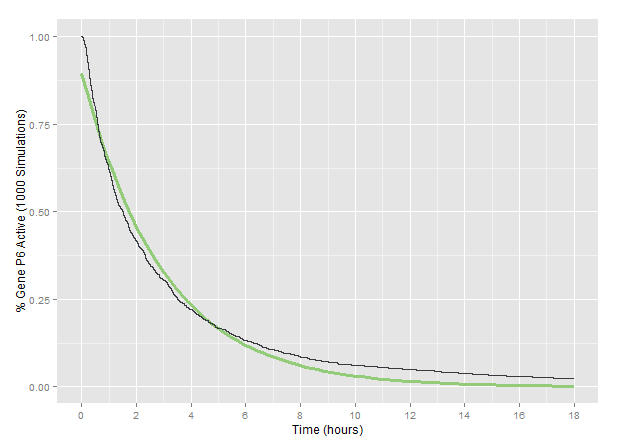
Based on the fit to our 1000-simulation average, we considered our CRISPR Plant Defense system to render the P6 gene of CaMV non-functional according to an exponential decay with a decay constant of 6.36x10
Discussion