Team:UChicago/Notebook

UChicago Summer 2015 Lab Journal
Courage is not the absence
of fear, but rather the judgment that something is more important than fear.
The brave may not live forever, but the cautious do not live at all.
-- Eduard
Christoff Philippe Gerard Renaldi,
Prince of Genovia, The
Princess Diaries
Week 1:
Goals- take inventory, design
constructs and primers, test competent cells
6/15/15
Worked on primer design quiz questions.
Discussed shift of project direction with Justin based on recently published
paper (Chen, 2015): http://advances.sciencemag.org/content/1/5/e1500358.full
Took inventory, created excel sheet
online under Protocols folder “Genehackers 2015
Inventory”
Downloaded SnapGene
Viewer to work on plasmid constructs.
6/16/15
New idea for output system -create one
construct with SasA/RpaA as
activator of output molecule, another with LabA/RpaA as inhibitor (negative feedback loop) of output
molecule. Based on article (Taniguchi, 2010). Started design on constructs,
possibly 5 in total:
1. Read-out activator (KaiCEE-RFP, RpaA, SasA)
2. Read-out inhibitor (KaiCEA-RFP, RpaA, CikA)
3. Test gene (kaibc promoter, GFP)
4. Fusion SasA
5. Fusion KaiC-P
Last two based heavily on paper (Chen,
2015)
Met with Jennifer Moran -need to do
safety training before autoclaving liquids and other procedures, will
accomplish once more people are back.
To Discuss: Is it worth having
negative feedback regulator of RpaA/ output molecule?
6/17/15
Decided to use CikA
as negative regulator/inhibitor instead of Lab A. CikA
better characterized. Reviewed (Gutu. O’Shea, 2013).
6/18/15
Finished design on constructs 1, 2, 3
Deciding on RBS, perhaps need to use high efficiency promoters and lower
efficiency RBS. Justin will email kaibc/Kai analog
sequences. Decided on meeting 4pm Tuesday.
To Discuss: Specific RBS and promoter
strengths on different genes.
Started Testing for Competent Cells
*Used Justin/Rust Lab protocol instead
of iGEM/team protocol because did not have cmrR plates.
Labels:
- Plasmid of
Interest- PJ006, containing KaiABC under kaiA, and kaibc promoters,
Spec resistance, Conc= 100ng/uL
- Transformed
plate- PJ006 MC 6/18/15
Steps
1. Remove cells from freezer, incubate on ice
2. Add 1 uL DNA (100ng/uL)
into competent cell tube
3. Incubate tube on ice for 30 mins
4. Incubate tube 42C water bath for 1 min heat shock
5. Incubate tube ice 5 mins
6. Rescue cells by pipetting 900 uL LB into
tube (use sterile flame)
*Used LB
from MJR Lab, will have to make LB tomorrow
7.
Incubate tube in shaker 37C for 1 hour
8.
Heat Spec plate in incubator as cold
plate reduces efficiency,complete while cells shaking
9.
Collect pellet, spin 3000 rcf/gs for 3 mins
10.
Decant 800 uL
of supernatant
11.
Use glass beads (5-6) per section (use
sterile flame)
12.
Mix pellet, pipetted 200 uL in total, 180 to 0.90 section, 20 to 0.10 section
13.
Shake with beads and remove
14.
Incubate plate overnight 37C
6/19/15
Checked on Transformed Plates
Good growth on both sections.
Transformation efficiency
0.10 Section
=
(293 cfus) / ( ((1 uL x 100 ng/uL)/1000 uL soln)(20/200 uL plated))
=
293 cfus/0.01 ng DNA plated = 2.93 x 10^4 transformants/ng
To Discuss: How to improve
Transformation Efficiency.
Made 500 mL LB Solution
Made CM plates
Temperature
of Freezer= 6 C, Chloramphenicol storage temperature= 2-8 C
125 uL of 50mg/ml cm used for every 250 uL
plate soln made
Poured
plates
Need
to label once plates set overnight
Updated restock list, will go tomorrow
to buy new supplies (check Inventory)
6/20/15
Stock room closed :(, Will order things
on Monday
Placed cmR
plates in left cold room, bottom right corner of room behind some Rust plates
Week 2:
Goals- Finalize construct design,
develop Competent Cells
6/22/15
Worked on Week 1 Presentation
Streaked 1 Tube of Comp 2014 Cells on
LB Only Plate for Competent E.coli procedure -protocol under master list
titled “Competent E.Coli 6/21/15”
Started Mini-prep to extract Kai
proteins. Inoculated 2 colonies, 1 each in 2mL LB + Spec medium.
6/23/15
Cultured 5 colonies from LB only plate
into 10 mL LB
Week 1 meeting today
Meeting Notes:
→ Discussed project direction and construct design. Need to add
terminator after RpaA. Need to decide whether or not
fusion protein construct worth it. Given that RpaA
might have some basal phosphorylation level, induced CikA
should present some results. How the constructs are set up now, ideal tests can
only be conducted with all three constructs. Don’t need to perhaps mutagenize
cut sites as these plasmids will not be final bio-brick. Final biobrick would possibly only have CikA,
SasA. Overall consensus is that CikA
worth exploring. Need to develop primers ASAP.
→ Need to develop more work on biosynthesis pathway and decide
which molecule want to consider as well as what is focus of experiment. Want KaiABC as submitted biobrick so
perhaps focusing on biosynthesis pathway is a bit ambitious. Need to consider
perhaps alternative, simpler molecule.
→ Will likely assemble using Gibson, order
this free kit from RPI.
Contact Danny for competent cells and
Justin for Gibson primers
6/24/15
Worked on designing primers. Contacted
Danny for competent cell procedure, will complete on Friday. Will conduct both
CaCl2 and RbCl2 procedures. Already have streaked LB only plate for competent
cells in cold room.
6/25/15
Finished designing primers, will check
with Kevin tomorrow. Inoculated two 5mL LB broths with 1-3 colonies each.
NEB Gibson Assembly Kit with competent cells arrived!
6/26/15
Carried out competent cell procedure.
https://drive.google.com/open?id=0B7wkycR1BRlmWHQ5UGNOaW0xX25ndmE1aUxPU01uclJCVmNV
Used both Rust Lab and RbCl2 as a
comparison.
Week 3:
Goals- Test efficiency of competent
cells, start as much cloning as possible
6/29/15
Conducted Transformation of CaCl2 and
RbCl2.
15.
Remove cells from freezer, incubate
tubes on ice
16.
Add 1 uL DNA
(50 pg//uL) into competent
cell tube
17.
Incubate tube on ice for 30 mins
18.
Incubate tube 42C water bath for 1 min
heat shock
19.
Incubate tube ice 5 mins
20.
Rescue cells by pipetting 850 uL LB into tube (use sterile flame)
21.
Incubate tube in shaker 37C for 1 hour
22.
Heat Cam plate in incubator as cold
plate reduces efficiency,complete while cells shaking
23.
Collect pellet, spin 3000 rcf/gs for 3 mins
24.
Decant 800 uL
of supernatant
25.
Use glass beads (5-6) per section (use
sterile flame)
26.
Mix pellet, pipetted 200 uL in total
27.
Shake with beads and remove
28.
Incubate plate overnight 37C
Spec on LB negative control culture
overnight =-0.019 A (no growth at all)
= (52 cfus) / ( ((1 uL
x 50 pg/uL x
1ng/1000pg)/1000uL soln))*((180/200 uL plated))
=
52 cfus/(4.5 x 10^-5) ng DNA
plated =1.15 x 10^6 transformants/ng (Rust)
=52 cfus) / ( ((1 uL
x 50 pg/uL x 1ng/1000pg)/1000uL
soln))*((180/200 uL
plated))
=
18 cfus/(4.5 x 10^-5) ng DNA
plated =4.00 x 10^5 transformants/ng (RbCl2)
6/30/15
Primers for first constructs arrived,
however at team meeting discussed how plasmids need to be re-designed to effectively
compare SasA and CikA. Also
CikA will need KaiB, and
likely to add RpaB to be consistent with Chen et al.
Constructs for Read-Out system were revamped, and constructs for Oscillation
system were designed as well.
Week 4:
Goals- Order final gBlocks and primers. Standardize and develop specific, in
depth protocols. Practice Western Blots, start writing project report.
7/20/15
gBlocks for Oscillation and Read-Out systems were modified. See Dropbox
for final edits and modifications.
7/21/15
gBlocks for Oscillation and Read-Out systems were finally ordered.
Primers were designed.
7/22/15
Primers ordered. Reached out to grad
advisers for Western Blotting techniques. Researched Gibson Assembly and
Western Blot protocols. Will need to research GFP Protocols.
GFP Protocols
http://advances.sciencemag.org/content/advances/1/5/e1500358.full.pdf
7/23
Materials for practice western blot
acquired. Gibson Assembly protocol drafted.
7/24
Started western blot. See Rust Lab
protocol. Slight changes include 7.5% gel used, cassette assembled not
submerged in buffer. Primers diluted and placed in -20 fridge.
7/27
Primary and secondary antibody staining
accomplished. Experiments more clearly laid out. Need to start considering plan
for pRha inducible promoter and how to alter
stoichiometry.
Week 5:
Goals- Finalize and outline
protocols, generate explanations for plasmids and background info for wiki and
presentation, order materials, gblock assembly
7/28
Western blot procedures expanded. See
Aaron’s email about compatible backbones. This week discussed assays -will need
to western blot for KaiA before investigating
oscillatory system in order to characterize input L-Rhamnose
to output Kai A production. Dilutions will occur on log scale first for L-Rhamnose.
7/29
Finished specific protocols -need to
ask White lab for sonicator?.
Looked up compatibility of backbones. pSB1,3,4 have
pMB1 (copy number 100-300/cell), p15A (low-medium 10-12 copy), pSC101 (~5
copies/cell). Will need to construct primers for kaibc/GFP
onto the SasA+CikA/SasA
plasmids.
7/30/15
Materials reviewed and listed. Meeting
with Barry to talk about iGEM as a class. Went over
protocols and methods. Allocated who is ordering what.
7/31/15
Gibson Assembly
5 uL of
Gibson HiFi Master mix was used in each assembly
reaction to minimize amount reagent used. Amount of blocks used, dependent on
bps of each block relative to each other. Each gblock
diluted in 20 uL of dH2O. Used
standardized amount 50 ng of largest block in each assembly. Assembled on ice.
Incubated on 50oC heatblock for 1 hour.
PCR
Assembled 5.5 times of 1X Master Mix
(not on ice). Dilute 100 uM (100X) primers to 10X
primers. 2 uL of primer and 18 uL
of dH2O. Aliquoted 49 uL of Master Mix
with 1 uL from Gibson Assembly Mix.
Recipe Phusion
Master Mix:
o Phusion 5X GC Buffer = 10uL x 5.5 = 55 uL
o dNTPs 10 mM = 1 x 5.5 =5.5 uL
o F Primer MC003 10 uM = 2.5 x 5.5 =13.75 uL
o R Primer MC004 10 uM = 2.5 x 5.5= 13.75 uL
o Phusion DNAP = 0.5 x 5.5 =2.75 uL
o H2O = 32.5
x 178.75 uL
o DMSO = 1.5 x 5.5 =8.25
uL
*Should have added only 170.5 uL (31 x 5.5) –Mix slightly
more dilute
Thermocycler Settings: 30 cycles, 98o
for 30s, 98o for 10s, 65o for 30s, 72o for 1
min, 72o 7 min, Hold 4o
Week 6:
Goals- gblock
assembly and Transformation
8/3/15
Decided on backbones:
Oscillator
MC001 – Cmr (standard igem
backbone for submitted biobrick)
Readout SasA MC002 –Amp (theoretically want to use with MC001 if
successful) à need to add GFP + kai bc
Readout SasA/CikA MC003 –Amp (“ “) à need
to add GFP + kaibc
KaiC Variants MC004-7- Cmr (would
never use with MC001)
Agarose Gel Casting
Made 50 mL of 1% Agarose gel. General
procedure: added agarose and 1X TAE into ER flask, microwaved until boil, cool
under water, poured into tray, added 0.75uL EtBr for visualizing, inserted
combs, cool in cold room for 15-20 mins.
Gel Electrophoresis
Loaded 10uL of 6X loading dye into 50uL
samples. Loaded 10 uL of 1 kB Plus DNA Ladder from
Invitrogen. Seems to be issue w/amount of sample loaded –only 15-29 uL available. Run under 120V for 30 mins. Could be issue
with evaporation in thermocycler. Gel too big for tray. Yields of products
exist, however seems low. Issue with MC001 –no clear product visible. PCR
should be redone.
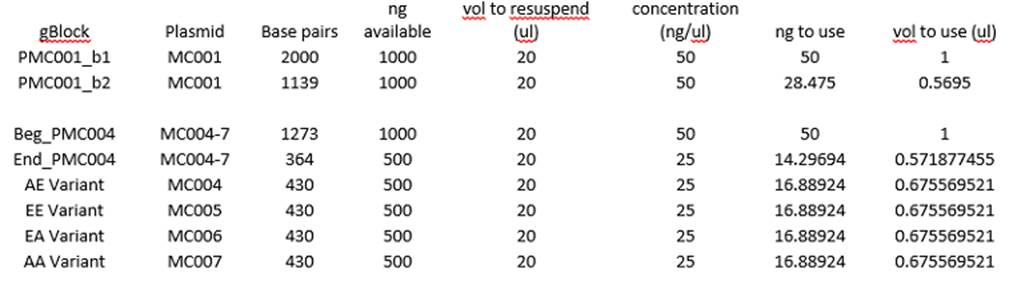
Image:
Gel Extraction
Gel Weights:
MC001 –N/A
MC004 -0.1536 g
MC005 -0.2048 g
MC006 -0.0965 g
MC007 -0.3962 g
*1mg = 1uL
Added 3x uL
QG buffer to volume of gel and 1X uL of isopropanol
to volume of gel.
PCR
To redo PCR for MC001,4,5,6,7,
1X Master Mix for 15 reactions made. Two reactions for each construct.
PCR Master Mix Recipe-
o Phusion 5X GC Buffer = 150 uL
o dNTPs 10 mM = 15 uL
o Phusion DNAP = 7.5 uL
o H2O = 465 uL
o DMSO = 22.5 uL
Added 2.5uL of F and R Primers, 44 uL of Master Mix, and 1uL of
DNA for each sample. Thermocycler settings- 98o 2 min, 98o
15 s, 69o 30 s, 72o 1 min, 72o 10 min, 4o
hold.
PCR Master mix
also used for amplifying linear Cmr backbones
(diluted in 10uL, used 1 uL of sample for Phusion PCR).
8/4/15
Gel Electrophoresis
Made 100 mL of 1% Agarose gel. 10 uL, 1kB plus Ladder loaded. 35uL MC001, 20uL MC001, 34 uL MC004, 34 uL MC004, 33 uL of MC006,7,8 and Cmr backbones. Products from MC004,5,6,7
and Cmr Linearized backbones extracted using gel
punches (borrowed from Rust Lab, need to order more to return). MC001 still not
very good yield. Next step to purify MC004-7 and linearized backbones, redo
Gibson and PCR of MC001 using gradient thermocycler.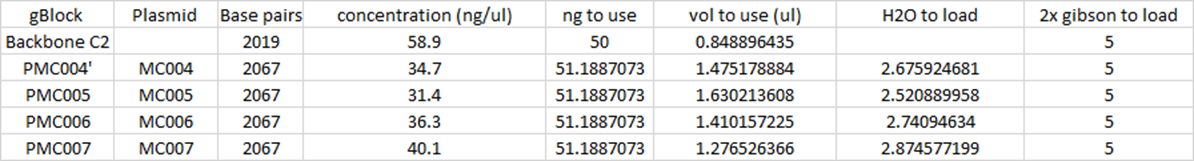
Image:
Gibson Assembly
5 uL of
Gibson HiFi Master mix, 1 uL
PMC001_b1, 0.5965 uL PMC001_b2, 3.4305 uL H2O, heatblock for
1 hour 50oC.
PCR
Master mix of
70 uL created (calculate ratio of 1X x 7/5)
o Phusion 5X HC Buffer = 14 uL
o dNTPs 10 mM = 1.4 uL
o Phusion DNAP = 0.7 uL
o H2O = 43.4 uL
o DMSO = 2.1 uL
o DNA =1.4 uL
o F Primer MC003= 3.5 uL
o R Primer MC004 =3.5 uL
10uL Master mix aliquoted into 7 samples.
Thermocycler settings- 98o 2
min, 98o 15 s, 60o, 62o, 64o,
66o, 68o, 70o, 72o 30 s, 72o
1 min, 72o 10 min, 4o hold.
Actual Anneal temperatures- 60.0o,
62.0o, 63.3o, 66.6o, 68.2o, 69.7o,
72.0o
T=66.0o, G=6.0o
for 30 cycles
8/5/15
Gel Electrophoresis
Made 90 mL of 1% Agarose gel. Loaded
10uL of 7 samples(+loading dye). Run for 120V, 30
mins. EtBr cloud on gel seen, only ladder shows visible bands. No other bands
visible. Likely error with PCR and addition of EtBr.
Image:
Gel Extraction
Used Promega
spin columns/buffer to concentrate in 15 uL of DNA
Purity Yields using nanodrop - C1 (Cam Backbone) -
C2 (Cam Backbone) -58.9 ng/uL
4 - 34.7
4’ -
5 - 31.5
6 - 36.3
7 - 40.1
PCR
To redo PCR for Gibson products of
MC001, 75 uL of 1X PCR Master Mix
PCR Master Mix Recipe-
o Phusion 5X HF Buffer = 15 uL
o dNTPs 10 mM = 15 uL
o Phusion DNAP = .75 uL
o H2O = 46.5 uL
o DMSO = 2.25 uL
o DNA (products from
Gibson 8/4)= 1.5uL
o F Primer MC003= 3.75uL
o R Primer MC004 =3.75uL
10 uL of
Master Mix aliquoted into each sample tube.
To PCR PMC001_b1 for confirmation of
block and analysis of primers
50 uL of
1XPCR Master Mix Recipe-
o Phusion 5X HF Buffer = 10 uL
o dNTPs 10 mM = 1 uL
o Phusion DNAP = .5 uL
o H2O= 31 uL
o DMSO = 1.5 uL
o DNA (pMC001_b1)= 1 uL
o F Primer MC005= 2.5uL
o R Primer MC006 =2.5uL
Added 2.5uL of F and R Primers, 44 uL of Master Mix, and 1uL of
DNA for each sample..
Thermocycler settings- 98o 2
min, 98o 15 s, 60o, 62o, 64o,
66o, 68o, 70o, 72o 30 s, 72o
1 min, 72o 10 min, 4o hold.
Actual Anneal temperatures- 60.0o,
62.0o, 63.3o, 66.6o, 68.2o, 69.7o,
72.0o
T=66.0o, G=6.0o
for 30 cycles
8/6/15
Gel Electrophoresis
Made 100 mL of 1% Agarose gel. Loaded
10uL of 7 samples(+loading dye) and 1 50uL sample
(divided into two wells, 42uL in one well 18 in the other). Run for 120V, 30
mins. No product clear enough to extract. Imaging gel shows faint products
under 68,70, and 72o. Could mean issue with
primers. Strangely, no product of right gBlock size
seen. Again could be primers.
Image:
Gibson Assembly:
Assembled purified biobricks
MC004, MC005, MC006, MC007 into Cam
backbone. Used following recipe based on bps of insert and backbone.

Transformation of Assembled products:
DNA straight from Gibson Assembly
Reaction was transformed into competent cells. RbCl2 competent cells used.
Efficiency of cells: 4.00 x 10^5 transformants/ng
(RbCl2)
1. Remove cells from freezer, incubate tubes on ice
2. Add 1 uL DNA (50 pg//uL) into competent cell tube
3. Incubate tube on ice for 30 mins
4. Incubate tube 42C water bath for 1 min heat shock
5. Incubate tube ice 5 mins
6. Rescue cells by pipetting 900 uL LB into
tube (use sterile flame)
7. Incubate tube in shaker at 37C/1100 RPM for 1 hour
8. Heat Cam plate in incubator as cold plate reduces efficiency,complete while cells shaking
9. Collect pellet, spin 3000 rcf/gs for 3 mins
10.
Decant 800 uL
of supernatant
11.
Use glass beads (5-6) per section (use
sterile flame)
12.
Mix pellet, pipetted 200 uL in total
13.
Shake with beads and remove
14.
Incubate plate overnight 37C

*Negative control w/no transformed DNA
resulted in 0 colonies. Negative control set up on 8/7.
PCR
PCR of 8/3 Gibson PCR, 8/4 Gibson PCR
conducted for further amplification. Block 4.1 PCR as positive control. Block
1.1 and 1.2 PCR run to increase DNA amount in hopes of Gibson from amplified
blocks. 50 uL of sample for each PCR (5 samples in
total).
Added Ingredients to individual Samples
o Phusion 5X HC Buffer = 10 uL
o dNTPs 10 mM = 1 uL
o Phusion DNAP = 0.5 uL
o H2O = 31.0 uL
o DMSO = 1.5 uL
o DNA =1.0 uL
o F Primer = 2.5 uL
o R Primer =2.5 uL
10uL Master mix aliquoted into 7 samples.
Thermocycler settings- 98o 2
min, 98o 15 s, 70o, 30 s, 72o 1 min, 72o
10 min, 4o hold.
Primers for each sample
Gibson 8/3 -MC003/MC004
Gibson 8/4 -MC003/MC004
pMC001_b1 -MC005/MC006
pMC001_b2 -MC007/MC008
pMC004_b1- MC019/MC020
*Upon further examination, should have
used MC003 for pMC004_b1
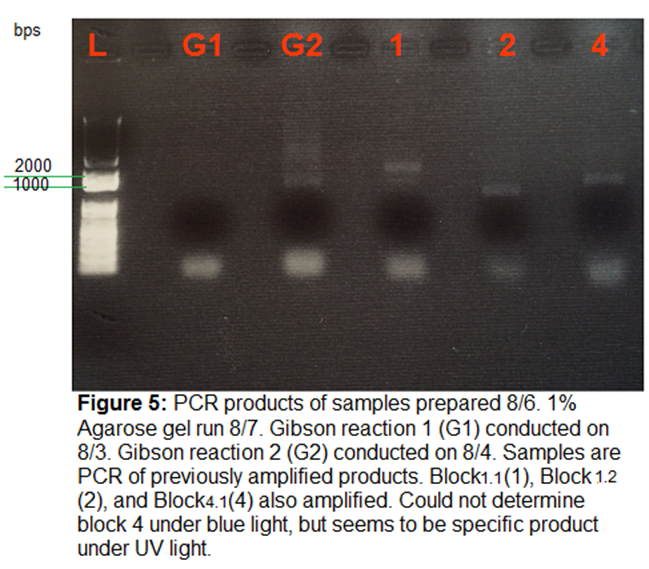
8/7/15
Gel Electrophoresis
1% Agarose gel run of PCR products from
8/6. Could not see products under blue light. Under UV light, products seemed
more specific. Still not as strong as in previous gels. Perhaps need to
troubleshoot PCR better.
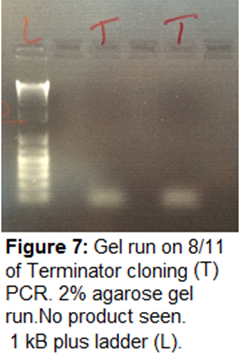
Image:
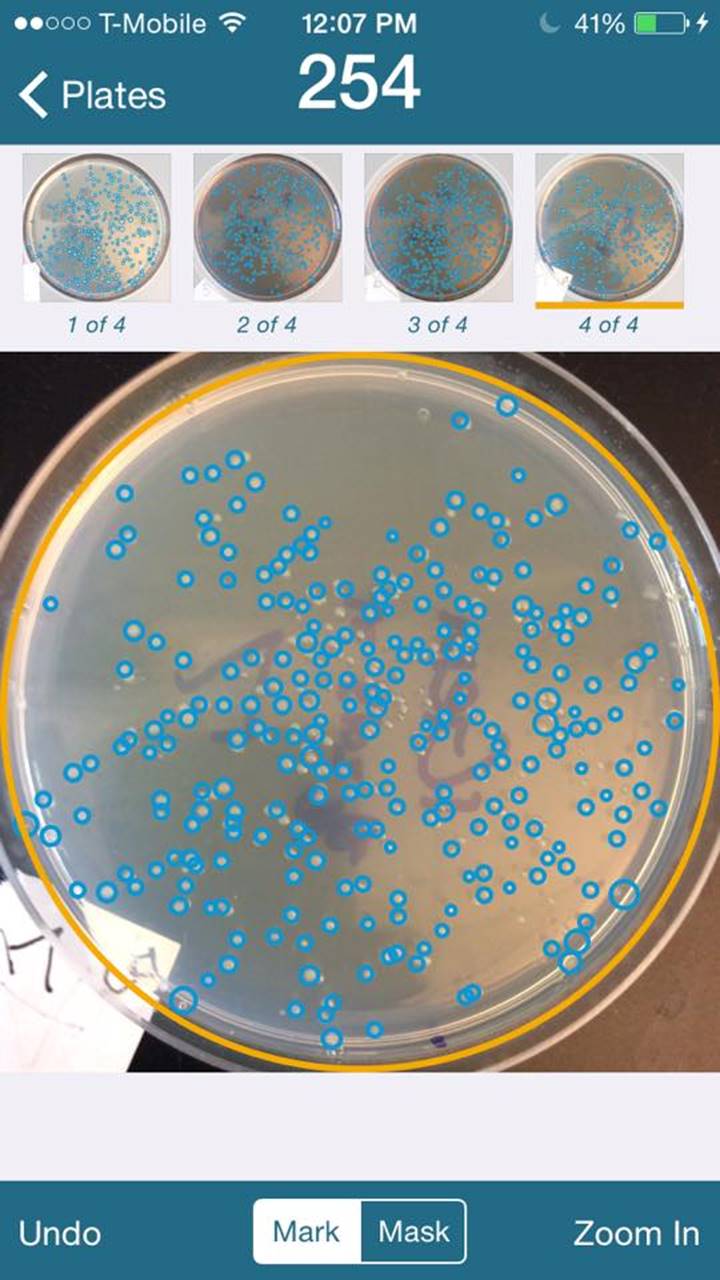
Transformation Results
Used iPhone application “ColonyCount” to assist in counting plates.
Plate Numbers are indicated on lower
left of the pictures.
Average is 321 colonies.


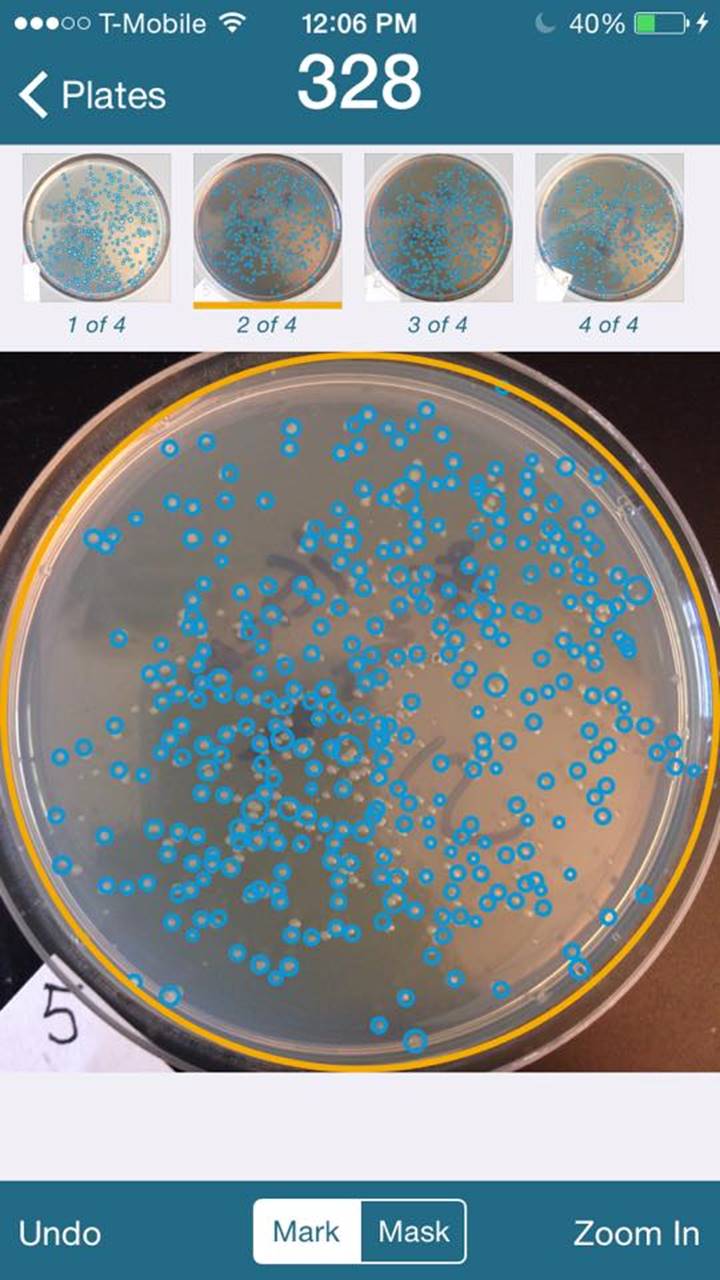

Transformation Efficiency
Used 1ul of 50pg/ul of DNA
4: 287 / 5*10^-5 = 5.74*10^6 cfu/ug
5: 328 / 5*10^-5 = 6.56*10^6 cfu/ug
6: 414 / 5*10^-5 = 8.28*10^6 cfu/ug
7: 254 / 5*10^-5 = 5.08*10^6 cfu/ug
Primers
Designed Sequencing primers as well as
new primers for pMC001. Primers made specifically for oscillator plasmid
-overhangs incorporated to make primers longer and more specific.
PCR
Prepared PCR of products seen on gel in
morning (G1, G2, 001b1, 001b2, 004b1, used 1 uL of
leftover PCR reaction). Used Q5 High Fidelity polymerase, as no Phusion available.
Ingredients:
Q5
High-Fidelity 2X Master Mix- 25 uL
DNA
-1 uL
F
Primer -2.5 uL
R
Primer -2.5 uL
H2O
-19 uL
Thermocycler Settings: 98C 30s, 98C
10s, 65C for 30s, 72C for 30s, 72C for 2mins, hold at 4C
30 cycles
Prepared Colony PCR To confirm inserts
of pMC004,5,6,7. Used Taq
DNA polymerase instead of phusion.
1. Pick single colony from plate, place in 50uL of dH2O (acts as DNA
template)
2. Add following PCR 1X Master Mix for Taq:
5 uL 10X buffer
1 uL dNTPs
1 uL 10 uM primer stock-VF2 and VR
1 uL DNA stock
0.5 uL Taq
41.5 uL H2O
Thermocycler Settings: 95C 2mins, 95C
15s, 55C 15s, 68C 45s (30 cycles), 68C
10m, hold 4C
-> Extend to 1min per kb (look up on
product sheet)
8/9/15
Gel Electrophoresis-
Ran 1% Agarose gel 120V, 30 mins. Ran
both Colony PCR and Q5 PCR. 5uL each sample loaded. Different ladder used
(Quick Load Purple 2-Log from NEB. Same amount of EtBr (0.75 uL) used.
8/10/15
Gel Electrophoresis-
Gel repeated, this time using leftover
45uL of sample. 1kB Plus invitrogen ladder used.
Innoculation-
Due to failure of Colony PCR, colonies
were inoculated and incubated. Will conduct direct miniprep
on 8/11 and sequence to sequence. This should help in determining if there is a
problem with primers/insert or with the PCR.
Gibson Assembly-
Gibson assembly of BH001_b1 and Cam backbone conducted.
PCR-
Set up PCR for biobricks
MC002, and MC003 and BH001 (confirmation).
11X PCR Master Mix
PCR Master Mix Recipe-
o Phusion 5X HF Buffer = 110 uL
o dNTPs 10 mM = 11 uL
o Phusion DNAP = 5.5 uL
o H2O = 341 uL
o DMSO = 16.5 uL
44uL of Master mix,
2.5 uL of F primer, 2.5 uL
of R Primer and 1 uL of template DNA used.
|
DNA Template |
Primers |
Info |
Gel to Run |
|
MC002_b1 |
MC029, MC010 1815
bps |
Clone
out b1 to give right initial sequence for kaibc/GFP/Term
inserts |
1%
Agarose |
|
MC003_b1 |
MC029, MC014 1815
bps |
Clone
out b1 to give right initial sequence for kaibc/GFP/Term
inserts |
1%
Agarose |
|
MC008_b1 |
MC003, MC028 1294
bps |
Clone
out kaibc promoter/GFP |
1%
Agarose |
|
Biobrick B0015
Terminator |
MC027, MC030 129
bps |
Clone
out terminator |
2%
Agarose |
|
BH001/Cam
Gibson |
MC003, MC004 765 bps *should
not have done |
?
Was to clone out insert, should have transformed as is. |
1%
Agarose |
Thermocycler settings- 98C 2min, 98C
15s, 67C 30s, 72C 1.5 mins, 72C 10min, 4 hold.
8/11/15
Gel Electrophoresis-
2% gel for Terminator cloning sample
and 1% gel for other PCR samples (see table) run. 120V for 30 min. 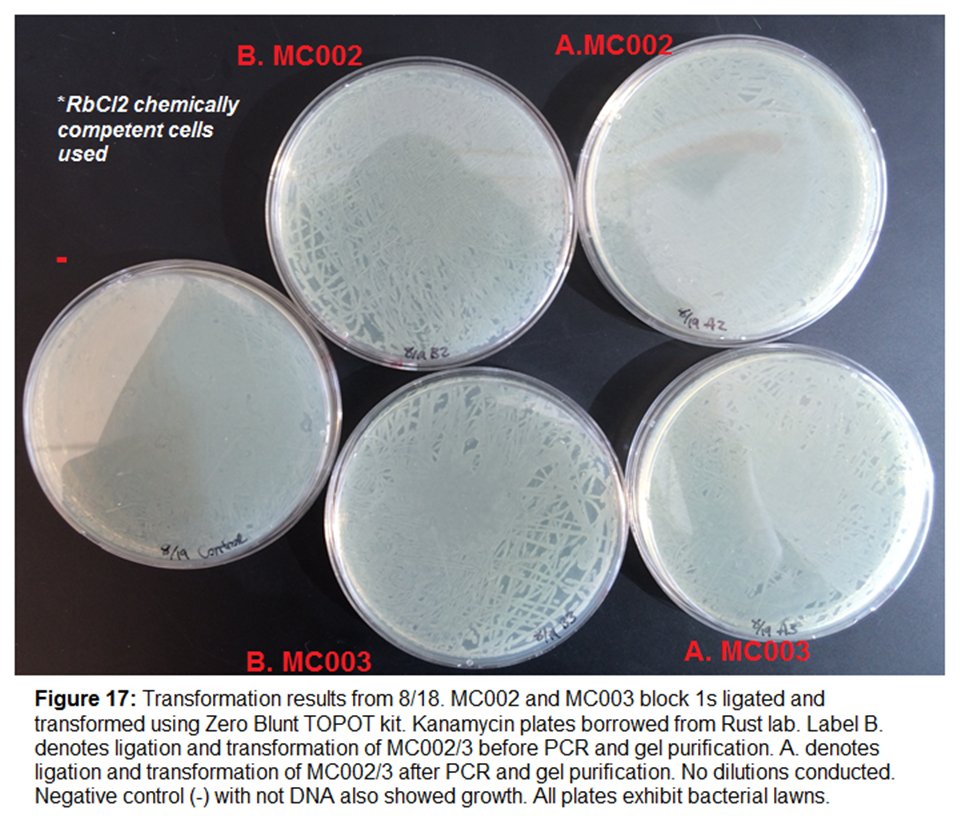

Miniprep
BH protocol
PCR
PCR of Minipreps
(BH protocol)
PCR
of MC002/3 blocks
Phusion 7 x of 1X mix
HF Buffer -70 uL
DMSO -10.5 uL
DNTPs -7 uL
H2O -219 uL
Phusion DNAP
-3.5 uL
Use 44 uL of
master mix w/ 2.5 of F and R primers and 1 uL of
template DNA.
|
DNA Template |
Primers |
|
MC002_b1 |
MC029, MC010 1815
bps product |
|
MC003_b1 |
MC029, MC014 1815
bps product |
|
MC008_b1 |
MC003, MC028 1294
bps product |
Thermocycler Settings-
|
STEP |
TEMP |
TIME |
|
Initial Denaturation |
98°C |
30 seconds |
|
30 Cycles |
98°C 65°C 72°C |
10 seconds 30 seconds 1 min (30s x 1.8kb -largest product) |
|
Final Extension |
72°C |
10 min |
|
Hold |
4°C |
Hold |
Thermocycler Settings for Mini-Prep
PCR-
|
STEP |
TEMP |
TIME |
|
Initial Denaturation |
98°C |
120 seconds |
|
30 Cycles |
98°C 61.6°C (NEB - DMSO%*0.8) 72°C |
15 seconds 30 seconds 1 min (30s x 1.8kb -largest product) |
|
Final Extension |
72°C |
5 min |
|
Hold |
4°C |
Hold |
8/12/15
PCR of Terminator
2 50 uL Samples
HF Buffer -10 uL
DMSO -1.5 uL
DNTPs -1 uL
H2O -31 uL
Phusion DNAP
-0.5 uL
10 uM of
MC027 Primer -2.5 uL
10 uM MC030
Primer -2.5 uL
DNA from plate -1 uL
Thermocycler
Settings: 1 cycle: 98C for 2 mins, 5 cycles: 98C for 15s, 69C for 30s, 72C for
2 min 30 cycles: 98C for 15s, 72C for 1.5 min, 1 cycle 72C for 10 min, 4C hold.
Gel
Electrophoresis-
50 mL 2%
gel, 150 mL 1% gel, and 100 mL 1% gel run for Miniprep
PCR as well as MC002/3 clone parts PCR. 25 uL of
sample loaded for Terminator 2% gel. 19 uL sample loaded for 150mL Miniprep
gel. 45 uL of sample loaded for MC002/3 clone parts
1% gel. 1kB plus Invitrogen ladder used. Send samples 41,43,51,52,53,54,61,62,63,64,74
for sequencing. 
Images:

Gel Purification-
Extracted correctly sized product fragmnets under blue light: T-189 bps, GFP 1249 bps.
Weights of gels:
GFP 1-63.8 mg
GFP 2-70.64 mg
T 1-34.6 mg
T 2-110.0 mg
Used Machery-Nagel
clean up method:
1. Add 200uL NTI Binding buffer / 100 mg gel
2. Incubate at 50C for 10 mins
3. Transfer solution to spin column and collection tube.
4. Spin at 11,000g (RCFs) for 30s
5. Discard flow through. Add 700 uL of wash
buffer NT3 to spin column.
6. Spin at 11,000g (RCFs) for 30s
7. Discard flow through. Add 700 uL of wash
buffer NT3 to spin column.
8. Spin at 11,000g (RCFs) for 30s
9. Spin at 11,000g (RCFs) for 1 min to dry silica membrane
10.
Elute DNA with 15 uL
of NE elution buffer. Spin at 11,000g (RCFs) for 30s
11.
Nanodrop (use EB buffer as blank, load 1.5 uL
sample)
Purity recorded w/Nanodrop:
T1- 5.1ng/uL
260/280-14.84
T2-23.1ng/uL
260/280-1.92
GFP 1- 41.6ng/uL
260/280-1.94
GFP 2- 33.1ng/uL
260/280-2.03
Plan for MC002_b1, MC003_b1: There is low yield of insert probably due to repeating regions of
DNA in blocks 1 of MC002 and MC003. As IDT expressed, there are a lot of low
mass products that are more efficiency amplified by primers. Therefore, as
Jennifer Moran mentioned, the best course of action would be to insert these
two gblocks into a Zero Blunt TOPO PCR Vector and
transforming into competent cells to amplify our gblocks.
After producing colonies, we can PCR and then screen for colonies with correct
product size. We will then miniprep and send these
blocks in for sequencing. This will take more time, but will ensure the purity
of the insert. After confirming the correct sequence, the DNA from the miniprep/gel extraction? can be
used to gibson the GFP/kaibc,
the terminator, and the first blocks together.
Mini-Prep Results (Nano-Drop)
|
Sample |
260/280 |
260/230 |
ng/ul |
|
41 |
1.86 |
2.20 |
55.1 |
|
42 |
1.84 |
2.06 |
35.4 |
|
43 |
1.89 |
2.10 |
72.9 |
|
44 |
1.92 |
1.83 |
30.7 |
|
51 |
1.94 |
1.87 |
46.8 |
|
52 |
1.90 |
2.00 |
53.9 |
|
53 |
1.90 |
2.15 |
66.6 |
|
54 |
1.93 |
2.14 |
58.6 |
|
61 |
1.97 |
2.06 |
52.2 |
|
62 |
1.97 |
2.14 |
61.1 |
|
63 |
2.00 |
2.24 |
52.5 |
|
64 |
1.92 |
2.12 |
57.1 |
|
71 |
2.03 |
2.13 |
56.9 |
|
72 |
1.88 |
1.81 |
50.2 |
|
73 |
1.93 |
1.83 |
59.3 |
|
74 |
1.92 |
1.92 |
53.3 |
The highlighted ones are the ones that
we choose to sequence.
Sequencing-
The concentration of the DNA templates
were too low, so we used 10 ug of each.
The primers were diluted to a 4uM
solution from a 100x stock.
(1.4 ul of
primer + 33.6 ul of water)
VF2 [1]
MC003 (F) [2]
MC041 (F) [3]
MC023 (F) [4]
MC022 (R) [5]
VR [6]
[41]
[43]
[53]
[54]
[62]
[63]
[71]
[73]

^Things sent.
Transformation of BH001:
DNA was transformed into competent
cells. RbCl2 competent cells used. Efficiency of cells: 4.00 x 10^5 transformants/ng (RbCl2)
15.
Remove cells from freezer, incubate
tubes on ice
16.
Add 1 uL DNA
(50 pg//uL) into competent
cell tube
17.
Incubate tube on ice for 30 mins
18.
Incubate tube 42C water bath for 1 min
heat shock
19.
Incubate tube ice 5 mins
20.
Rescue cells by pipetting 900 uL LB into tube (use sterile flame)
21.
Incubate tube in shaker at 37C/1100 RPM
for 1 hour
22.
Heat Cam plate in incubator as cold
plate reduces efficiency,complete while cells shaking
23.
Collect pellet, spin 3000 rcf/gs for 3 mins
24.
Decant 800 uL
of supernatant
25.
Use glass beads (5-6) per section (use
sterile flame)
26.
Mix pellet, pipetted 200 uL in total
27.
Shake with beads and remove
28.
Incubate plate overnight 37C
8/13/15-

Transformation Efficiency of BH001-
amt dna used/plate
Plates 1 and 2 were discarded, a colony
PCR was done on 6 samples from Plate 3
They were PCRed
separately with two different annealing temperatures, 61.6 and 66.
Gibson Assembly
Gibson assembly using 5uL of gibson master mix 2x conducted.
Assembled on ice. Incubated for 1 hour 50C heatblock.

PCR-
Prepared 2 50uL 1X PCR samples.
HF Buffer -10 uL
DMSO -1.5 uL
DNTPs -1 uL
H2O -31 uL
Phusion DNAP
-0.5 uL
F Primer 10 uM
MC003 -2.5 uL
R Primer 10uM MC004 -2.5 uL
DNA (Gibson assembly rxn 8/13) -1uL
Thermocycler Settings-
|
STEP |
TEMP |
TIME |
|
Initial Denaturation |
98°C |
30 seconds |
|
30 Cycles |
98°C 65°C 72°C |
10 seconds 30 secondsc 1.65 mins (30s x 3.3kb -largest product) |
|
Final Extension |
72°C |
10 min |
|
Hold |
4°C |
Hold |
Thermocycler Settings for Mini-Prep
PCR- Low Temp
|
STEP |
TEMP |
TIME |
|
Initial Denaturation |
98°C |
120 seconds |
|
30 Cycles |
98°C 61.6°C (NEB - DMSO%*0.8) 72°C |
15 seconds 30 seconds 1 min (30s x 1.8kb -largest product) |
|
Final Extension |
72°C |
5 min |
|
Hold |
4°C |
Hold |
Thermocycler Settings for Mini-Prep
PCR- High Temp
|
STEP |
TEMP |
TIME |
|
Initial Denaturation |
98°C |
120 seconds |
|
30 Cycles |
98°C 66°C 72°C |
15 seconds 30 seconds 1 min (30s x 1.8kb -largest product) |
|
Final Extension |
72°C |
5 min |
|
Hold |
4°C |
Hold |
Gel Electrophoresis-
100 mL of 1% agarose gel run 120V, 30 mins. Samples loaded include
45 uL of BH003 biobrick and
20 uL of colony PCRs. 5 uL
1 kB plus Invitrogen ladder loaded. Colony PCRs yielded no results, biobrick BH003 extracted using gel punches (seen in image).
First column of BH3 seemed to give very low yield (light band not strong band
seen before extraction).
Image-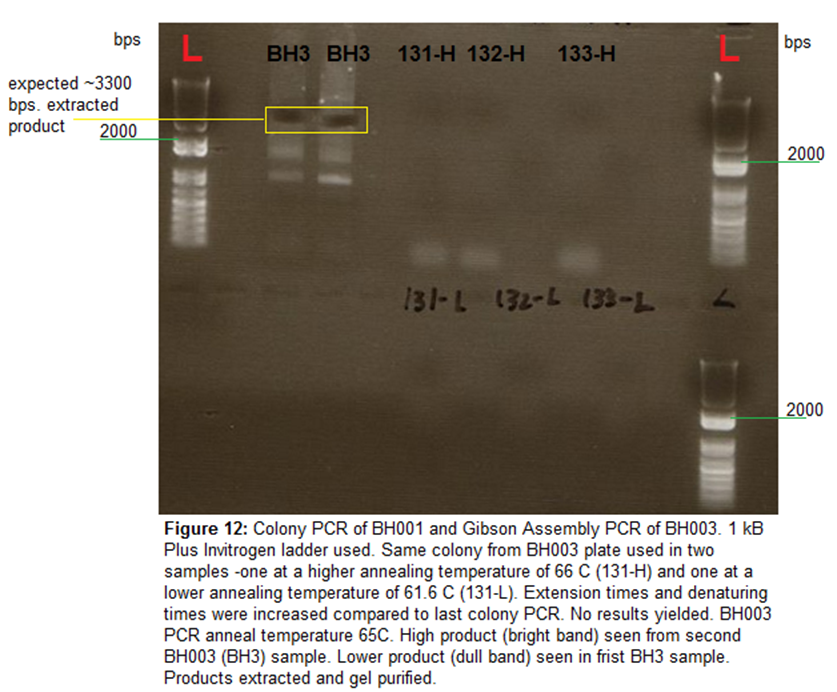
Gel Extraction-
BH003 biobrick
extracted from gel. Machery-Nagel protocol used for
gel extraction.
Weight of gel: 85.2 mg
Used Machery-Nagel
clean up method:
12.
Add 200uL NTI Binding buffer / 100 mg
gel
13.
Incubate at 50C for 10 mins
14.
Transfer solution to spin column and
collection tube.
15.
Spin at 11,000g (RCFs) for 30s
16.
Discard flow through. Add 700 uL of wash buffer NT3 to spin column.
17.
Spin at 11,000g (RCFs) for 30s
18.
Discard flow through. Add 700 uL of wash buffer NT3 to spin column.
19.
Spin at 11,000g (RCFs) for 30s
20.
Spin at 11,000g (RCFs) for 1 min to dry
silica membrane
21.
Elute DNA with 15 uL
of NE elution buffer. Spin at 11,000g (RCFs) for 30s
22.
Nanodrop (use EB buffer as blank, load 1.5 uL
sample)
Nanodrop purity- 14.4 ng/uL, 260/280- 1.55
PCR-
Made 4 1X 50uL samples to PCR ampicillin backbone. Used 1 uL of 25ng/uL amp linearized
backbone. Primers MC001 and MC002.
PCR Master Mix Recipe-
o Phusion 5X HF Buffer = 50 uL
o dNTPs 10 mM = 5 uL
o Phusion DNAP = 2.5 uL
o H2O = 155 uL
o DMSO = 7.5 uL
o DNA (products from
Gibson 8/4)= 1.0uL
o F Primer MC001= 2.5uL
o R Primer MC002 =2.55uL
Thermocycler settings:98C
30s, 25 cycles: 98C 10s, 69C 30s, 72C 1.5m, 72.0C 10 min, 4C hold
Gibson Assembly-
Given success of gibson
and transformation with BH001, decided to try assemble pMC001 and transform
directly. Reaction incubated on heatblock for 1 hour
at 50C.

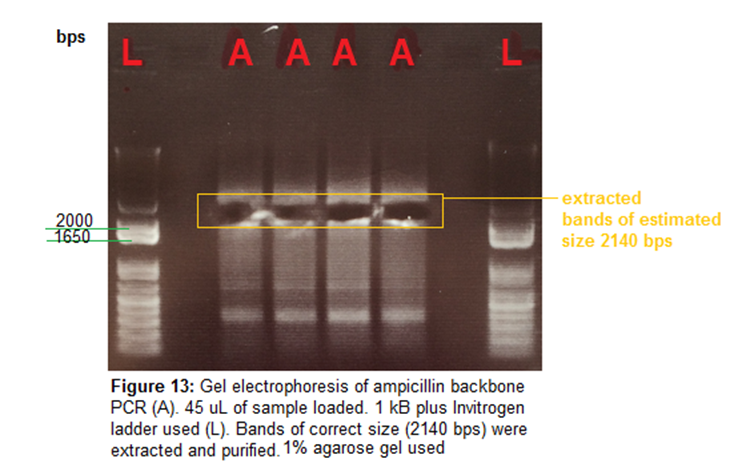
Gel Electrophoresis-
50 mL of 1% agarose gel made to run amp backbone amplifications.
0.5 uL EtBr used. Gel run 120V for 30 mins. 1 kB Plus
Invitrogen ladder used. 45 uL of samples loaded.
Image- 
Gel Extraction and Purification-
Ampicillin backbone
extracted from gel. Machery-Nagel
protocol used for gel extraction.
Weight of gel samples:
A1 -135.8 mg (271.6 uL
of binding buffer NTI used)
A2 -201.4mg (402.8 uL
of binding buffer NTI used)
Nanodrop concentrations of ampicillin backbone samples
A1 - 32.3 ng/uL,
260/280- 1.84
A2 - 33.4 ng/uL,
260/280- 1.59
Gibson-
Gibson assembly of ampicillin backbone
to BH003 insert conducted. Used 5uL of gibson
master mix 2x conducted. Assembled on ice. Incubated for 1 hour 50C heatblock.
Transformation of BH003:
DNA was transformed into competent
cells. RbCl2 competent cells used. Efficiency of cells: 4.00 x 10^5 transformants/ng (RbCl2)
29.
Remove cells from freezer, incubate
tubes on ice
30.
Add 1 uL DNA
(50 pg//uL) into competent
cell tube
31.
Incubate tube on ice for 30 mins
32.
Incubate tube 42C water bath for 1 min
heat shock
33.
Incubate tube ice 5 mins
34.
Rescue cells by pipetting 900 uL LB into tube (use sterile flame)
35.
Incubate tube in shaker at 37C/1100 RPM
for 1 hour
36.
Heat Cam plate in incubator as cold
plate reduces efficiency,complete while cells shaking
37.
Collect pellet, spin 3000 rcf/gs for 3 mins
38.
Decant 800 uL
of supernatant
39.
Use glass beads (5-6) per section (use
sterile flame)
40.
Mix pellet, pipetted 200 uL in total
41.
Shake with beads and remove
42.
Incubate plate overnight 37C
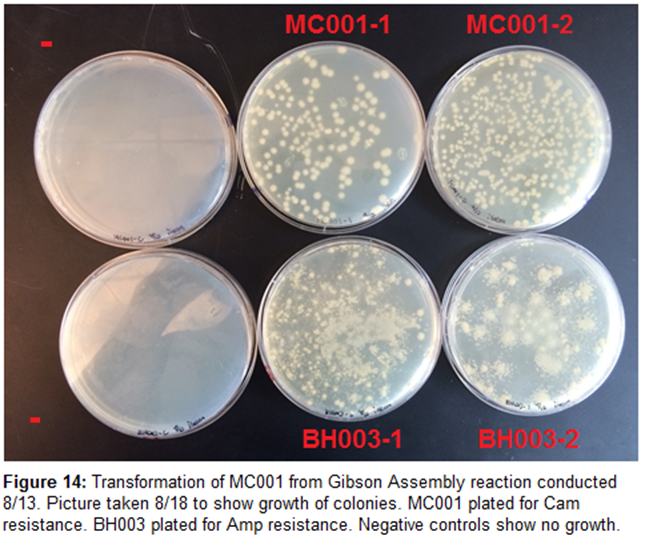
8/14/15
Transformation-
MC001
and BH003 gave good results from transformation. Colonies were small, but
evenly distributed. Colonies could be small because plates incubated late last
night.

Colony PCR of BH003/MC001-
Diluted one colony from plates with
most growth into 50uL of dH2O to serve as DNA template.
Made 8.5 x of 1X PCR Master Mix:
HF
Buffer: 85 uL
DMSO:
12.75 uL
dNTPS: 8.5 uL
H2O:
263.5 uL
DNAP:
4.25 uL
Used
2.5uL of VF2 and VR each and 1 uL of sample DNA.
Thermocycler Settings for Colony
|
STEP |
TEMP |
TIME |
|
Initial Denaturation |
98°C |
10 minutes |
|
30 Cycles |
98°C 66°C 72°C |
15 seconds 30 seconds 2.5 mins |
|
Final Extension |
72°C |
10 min |
|
Hold |
4°C |
Hold |
Sequencing Primers for MC001- MC003,MC031,MC032,MC006,MC033, MC008
8/15/
Gel Electrophoresis
1% Agarose gel run for colony PCR. 1 kB Invitrogen plus ladder
used. 50 uL of sample loaded.

8/17
Sequences Submitted
MC001 note submitted on weekend for
sequencing. MC001 submitted in morning for sequencing. 4uM primer stock made by
diluting in water (1:24 primer:water
ratio). 14 uL of miniprepped
DNA submitted with 10uL of 4uM primers.
Primers used:
VF2 (1)
VR (2)
MC003 (3)
MC031 (4)
MC032 (5)
MC006 (6)
MC033 (7)
MC008 (8)
Colony PCR
Colony PCR of MC001 set up again as
upon closer examination, insert seems too small to be correct.
Diluted one colony from plates with
most growth into 50uL of dH2O to serve as DNA template.
Made 9/5x of 1X PCR Master Mix for 10uL
samples (used 9.8 uL Mix and 0.2 uL
DNA):
HF
Buffer: 18.00
DMSO:
2.70 uL
dNTPS: 1.80
H2O:
55.
DNAP:
4.25 uL
Used
4.50 uL of 10X MC003 and MC004 for mix.
Thermocycler Settings for Colony
|
STEP |
TEMP |
TIME |
|
Initial Denaturation |
98°C |
10 minutes |
|
30 Cycles |
98°C 66°C 72°C |
15 seconds 30 seconds 2.5 mins |
|
Final Extension |
72°C |
10 min |
|
Hold |
4°C |
Hold |
Talked with Justin about ordering
materials -TOPO kit should come tomorrow.
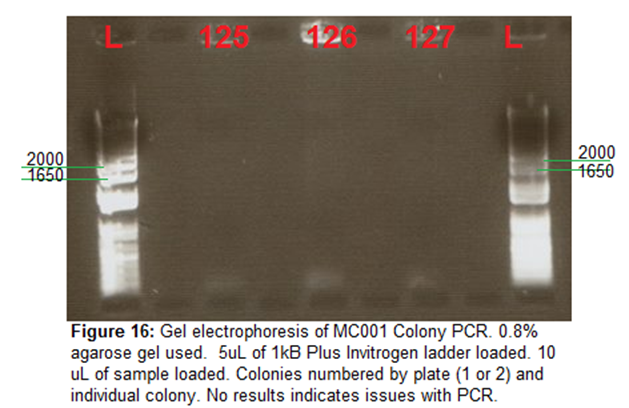
8/18
Gel Electrophoresis-
0.8% (to separate out larger fragments
with more efficiency) agarose gel cast. Gel run on 120V for 30 mins. 1kB plus
Invitrogen ladder used. No results from colony PCR, indicating something wrong
during PCR. Likely issue with primers? ¾ Used instead of VF2 and VR. Have
double checked primers are correct -could be issue with insert and primers.
Note -issues with evaporation in thermocycler, which is why samples
115,116,117,118,128 could not be loaded.
Image-

Sequencing Results
Sequencing successful for MC004,5,7. MC006 and MC007 samples probably mislabelled
as sequencing indicates correct MC006 phosphomimetic
is in MC007 sequence and vice versa. Otherwise, MC007,5,
and 4 are all correctly sequenced. Point mutation in MC006 (labelled MC007) samples
base 244 of Snapgene file. Point mutation seems to be
silent (GGC to GGT, translate features lists both as glycine. Four read out KaiC variants seem good to go.
MC001 -still waiting for colony PCR/
specific primers to arrive for assembly
MC002/3 -still waiting for TOPO kit to
come for transformation ligation and assembly
MC004/7 -all assembled
BH001 -assembled, sequence verify
BH002 -gblocks
not arrived yet
BH003 -assembled, sequence verify
BH004 -gblocks
not arrived
Zero-Blunt TOPT Ligation and Transformation
We will be ligating and transforming four samples in order to
allow the bacteria to amplify blocks 1 of MC002 and MC003.
- MC002_b1
- MC003_b1
- MC002_b1 after
PCR
- MC003_b1 after
PCR
Ligation
1. Combine 2uL product, 1 uL provided salt
solution from pTOPO kit, 2uL H20, 1 uL pCR II-Blunt-TOPO vector
2. Pipette up and down a few times to mix.
3. Incubate 5 min at room temperature.
4. No overnight incubation needed! But you can leave it
overnight at 4°C if you cannot proceed with the transformation right away,
Transformation
1. Thaw one vial of Chemically Competent E. coli cells per
transformation on ice.
2. Add 2μl ligation product to the thawed cells. Keep on
ice for 30 mins.
3. Transfer vials to a 42°C water bath for 45 seconds. Immediately
return the tubes to ice for 2 min.
4. Add 250μl room temperature SOC orLB
to each vial.
5. Incubate for 1 hr at 37°C.
6. Plate 200μl cells on LB + kanamycin plates.
7. Incubate overnight at 37°C
*The vector confers resistance to
kanamycin, NOT ampicillin.
pTOPO kit)
Note: if you do not have a
sufficient number of tranformants in 200μl,
repeat the transformation, do a short spin (20‐30 sec) to
gently pellet your cells just before plating. Remove 100μl of the
supernatant, resuspend the cells by pipeting up and down.
PCR
Set up PCR
for amplifying MC002_b1 and MC003_b1. The products from this amplification
(correct size ~1850-1970 bps) will be ligated and transformed using the TOPO
zero blunt kit in order to have more specific inserts.
Set
up 2 50uL 1X PCR Reactions:
- dNTPS: 1uL
- H2O: 31 uL
- DNAP: 0.5 uL
- DNA: 1 uL (gblocks MC002_b1 and
MC003_b1)
- DMSO: 1.5uL
- HF Buffer: 10uL
- F Primer: 2.5uL
(MC029)
- R Primer: 2.5 uL (MC010 for MC002_b1 and MC014 for MC003_b1)
Thermocycler settings for gBlock PCR
|
STEP |
TEMP |
TIME |
|
Initial Denaturation |
98°C |
30 seconds |
|
30 Cycles |
98°C 66°C 72°C |
10 seconds 30 seconds 1.65 mins |
|
Final Extension |
72°C |
10 min |
|
Hold |
4°C |
Hold |
Innoculation
Colonies from MC001 plates were inoculated as colony PCR results
were ambiguous. Samples: 115,116,117,118,125,126,127,128.
1. Aliquot enough LB for 2mL/sample into a Falcon tube. Pipette 2mL
of LB into each culture tube/sample.
2. Pipette all of the colony suspension into the culture tube as
well.
3. Put in antibiotic to act as primary screening for insert -Cam 1uL,
Amp 4uL (antibiotics stored in the -20 fridge)
4. Place culture tubes into shaker at 37C overnight (~16 hrs)
Gel Electrophoresis
1% agarose gel used to run gel electrophoresis for MC002_b1 and
MC003_b1. Gel run under 120V for 30 mins. Gel extraction of estimated product
size conducted under blue light. 5uL 1kB Plus Invitrogen ladder loaded. 50uL of
samples loaded.
Image
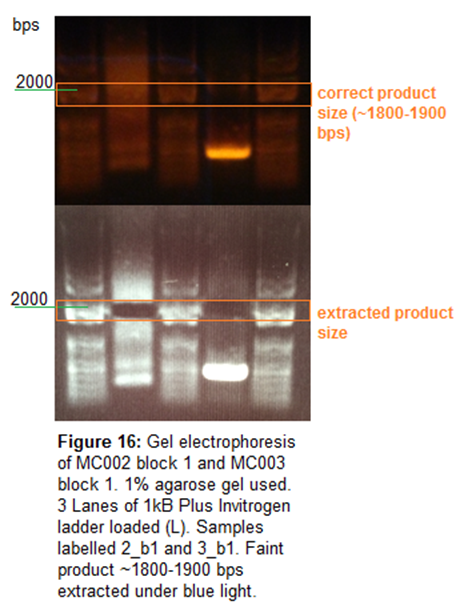
8/19/15

Transformation Results
Bacterial lawns seen on all plates. Perhaps issue with plates or
competent cells are naturally resistant to kanamycin? Will try dilutions if
cells are very efficient as cells could also be very conducive to
transformation.
Sequencing Results
Analyzed samples 124 and 121 using Blast and chromatograms. Both
samples did not have whole insert -MC031 gave no results for both samples. In
both samples sequencing only at end of insert (part of KaiC
w/KaiB and suffix) resulted.
Miniprep-
Miniprep inoculations 115,116,117,118,125,126,127,128.
1. Centrifuge cells at 12000 rcf for 3 mins
to harvest cells. Remove all medium.
2. Resuspend cells by adding 250uL of resuspension buffer R3 with RNase A. Mix
up and down until homogenous.
3. Add 250uL of Lysis buffer (L7) to lyse cells. Mix gently by
inverting capped tube.Do not vortex, incubate at room
temp for 5 mins.
4. Add 350uL of precipitation buffer N4. mix
immediately by inverting tube or vigorously shaking. Do not vortex, centrifuge
at 20,000 rcfs for 10 mins.
5. Load supernatant (750 uL) into Spin
column (machery nagel used)
and collection tube. Centrifuge column at 12,000 rcfs
for 1 min. Discard flow through and place column back in wash tube.
6. Add 500 uL of wash buffer W10 with
ethanol. Incubate at room temp for 1 min, centrifuge column at 12,000gs for 1
min. Discard flow through. (*Optional Wash step)
7. Add 700 uL of wash buffer W9 with
ethanol. centrifuge column at 12,000 gs for 1 min. Discard flow through then dry spin for 1 min
at 12,000 gs. Discard flow through.
8. Place spin column into microfuge tube. Add 50uL of TE Buffer to
center of column. Incubate column in room temp for 1 min.
9. Centrifuge column at 12,000 rcfs for 2
mins. Discard column. Nanodrop DNA. Store at 4C short
term, -20C long term.
Nanodrop results-
115: 91.8 ng/uL,
260/280: 1.90
116: 172.0 ng/uL,
260/280: 1.82
117: 152.0 ng/uL,
260/280: 1.91
118: 120.4 ng/uL,
260/280: 1.81
125: 187.0 ng/uL,
260/280: 1.88
126: 149.2 ng/uL,
260/280: 1.88
127: 211.0 ng/uL,
260/280: 1.87
128: 222.8 ng/uL,
260/280: 1.89
PCR
Set up PCR for both miniprep PCR and PCR
of MC002_b1 and MC003_b1 to redo ligation and transformation.
Set up 2 50uL 1X PCR Reactions for
MC002_b1 and MC003_b1:
- dNTPS: 1uL
- H2O: 31 uL
- DNAP: 0.5 uL
- DNA: 1 uL (gblocks MC002_b1 and
MC003_b1)
- DMSO: 1.5uL
- HF Buffer: 10uL
- F Primer: 2.5uL
(MC029)
- R Primer: 2.5 uL (MC010 for MC002_b1 and MC014 for MC003_b1)
Made 9/5x of 1X PCR Master Mix for 10uL
samples (used 9.8 uL Mix and 0.2 uL
DNA):
HF
Buffer: 18.00
DMSO:
2.70 uL
dNTPS: 1.80
H2O:
55.
DNAP:
4.25 uL
Used
4.50 uL of 10X MC003 and MC004 for mix.
Thermocycler Settings (used 2
thermocyclers, one for 10uL samples and other for 50uL samples)-
|
STEP |
TEMP |
TIME |
|
Initial Denaturation |
98°C |
30 seconds |
|
30 Cycles |
98°C 67°C 72°C |
10 seconds 30 seconds 1:40 mins |
|
Final Extension |
72°C |
10 min |
|
Hold |
4°C |
Hold |
Gel Electrophoresis-
1% agarose
gel run at 120V for 30 mins. Loaded 10uL and 45 uL
for minipreps and MC002/3 gel extractions
respectively. Loaded 5uL of 1kB Plus Invitrogen ladder.
Image-

8/20
Sequences Submitted
MC001 samples submitted in morning for
sequencing. 4uM primer stock made by diluting in water (1:24 primer:water ratio). 10 uL of miniprepped DNA submitted
with 10uL of 4uM primers. Samples 115,117, 125, 127, 128 sent.
Primers used:
VF2 (1)
MC003 (2)
MC031 (3)
MC032 (4)
MC006 (5)
MC033 (6)
MC008 (7)
VR (8)
Gibson Assembly
New primers for MC001 arrived.
Proceeded with Gibson assesmbly. Incubated mix at 50C
for 1 hour.
PCR
Set up PCR for MC002_b1, MC003_b1,
MC001_b1, MC001_b2, and MC001 Gibson products. Total 7 reaction (MC002/3 block
1s sampled twice as one will be used for gel extraction then TOPO
ligation/transformation, the other will be used to amplify a second time).
Phusion 7.5 x of 1X mix
HF Buffer -75.00 uL
DMSO -11.25 uL
DNTPs -7.50 uL
H2O -232.50 uL
Phusion DNAP
-3.75 uL
Add 44uL of PCR Mix to 2.5 of F and R
Primer each and 1 uL of DNA
DNA : Primers
MC002_b1 :
MC029, MC010
MC003_b1 :
MC029, MC014
Gibson rxn : 1b1F, 1b2R
MC001_b1 :
1b1F, 1b1R
MC002_b2: 1b2F, 1b2R
Thermocycler settings:
Thermocycler Settings (used 2
thermocyclers, one for 10uL samples and other for 50uL samples)-
|
STEP |
TEMP |
TIME |
|
Initial Denaturation |
98°C |
30 seconds |
|
30 Cycles |
98°C 66°C 72°C |
10 seconds 30 seconds 1:30 mins |
|
Final Extension |
72°C |
10 min |
|
Hold |
4°C |
Hold |
Gel Electrohporesis-
1% Agarose gel run at 120V for 30 mins. 45 uL
of samples loaded. Hard to see bands under blue light. Used UV light for quick
clarification of size. No products from MC001 Gibson seen. MC001_b1 and
MC001_b2 extracted separately.
8/21
PCR
Due to failure of yesterday’s PCR, gradient PCR was set up in
order to determine optimal anneal temperature.
Master mix of
75 uL created (calculate ratio of 1X x 7.5/5)
o Phusion 5X HF Buffer = 15 uL
o dNTPs 10 mM = 1.5 uL
o Phusion DNAP = 0.75 uL
o H2O = 46.5 uL
o DMSO = 2.25 uL
o DNA =1.5 uL
o F Primer MC003= 3.75 uL
o R Primer MC004 =3.75 uL
10uL Master mix aliquoted into 7
samples.
Thermocycler settings- 98o 2
min, 98o 15 s, 60o, 62o, 64o,
66o, 68o, 70o, 72o 30 s, 72o
1 min, 72o 10 min, 4o hold.
Actual Anneal temperatures- 60.0o,
62.0o, 63.3o, 66.6o, 68.2o, 69.7o,
72.0o
T=66.0o, G=6.0o
for 30 cycles
Gel Purification
MC001_b1 and MC001_b2 extracted yesterday were purified. Used Machery-Nagel protocol.
Weights-
1b1 -13.4 mg
1b2 - 133.4 mg
Nanodrop results-
1b1 - 13.1 ng/uL
1b2 -19.2 ng/ul
Gel Electrophoresis
1% agarose gel run for gradient PCR. 120V for 30 mins. 1kB
Invitrogen ladder used. 10uL of sample loaded. Expected product size ~3000 bps.
No expected product size appeared on gel. Perhaps indicates issues with Gibson
assembly. 24 inoculated colony PCRs also run on separate gel. 3 colonies
chosen for inoculation and miniprep: 3,9 and 22.
Image-


Gibson-
Gibson of MC001 repeated. Incubated mix at 50C for 1 hour.
PCR-
PCR set up for MC001 blocks and MC001 8/21 Gibson. 6 samples in
total. 1X PCR Mix for each sample set up:
DMAP:
0.5 uL
DMSO:
1.5 uL
dNTPS: 1 uL
DNA:
1uL
HF
Buffer: 10 uL
H2O:
31 uL
F
Primer: 2.5 uL (10uM)
R
Primer: 2.5 uL (10uM)
DNA (Primers): MC001_b1
(MC001_b1_F_new, MC001_b1_R_new), MC001_b1 (MC001_b2_F_new, MC001_b2_R_new),
MC001 Gibson (MC001_b1_F_new, MC001_b2_R_new)
PCR Conducted one at high anneal
temperature (68C) one at low anneal temperature (64C)
Thermocycler Settings (used 2 thermocyclers,
one for 10uL samples and other for 50uL samples)-
|
STEP |
TEMP |
TIME |
|
Initial Denaturation |
98°C |
30 seconds |
|
30 Cycles |
98°C 68 or 64°C 72°C |
10 seconds 30 seconds 1:30 mins |
|
Final Extension |
72°C |
10 min |
|
Hold |
4°C |
Hold |
Gel Electrophoresis-
1% agarose gel run at 120V for 30 mins. 1kB Plus invitrogen ladder used. 50uL of samples loaded. Results
very inconclusive.
Image-
8/24
Miniprep-
Inoculated
TOPO MC002_b1 and MC003_b1 samples miniprepped using invitrogen protocol.
Miniprep inoculations 115,116,117,118,125,126,127,128.
10.
Centrifuge cells at 12000 rcf for 3 mins to harvest cells. Remove all medium.
11.
Resuspend cells by adding 250uL of resuspension buffer R3 with RNase A. Mix
up and down until homogenous.
12.
Add 250uL of Lysis buffer (L7) to lyse
cells. Mix gently by inverting capped tube.Do not
vortex, incubate at room temp for 5 mins.
13.
Add 350uL of precipitation buffer N4. mix immediately by inverting tube or vigorously shaking. Do
not vortex, centrifuge at 20,000 rcfs for 10 mins.
14.
Load supernatant (750 uL) into Spin column (machery nagel used) and collection tube. Centrifuge column at
12,000 rcfs for 1 min. Discard flow through and place
column back in wash tube.
15.
Add 500 uL of
wash buffer W10 with ethanol. Incubate at room temp for 1 min, centrifuge
column at 12,000gs for 1 min. Discard flow through. (*Optional Wash step)
16.
Add 700 uL of
wash buffer W9 with ethanol. centrifuge column at
12,000 gs for 1 min. Discard flow through then dry
spin for 1 min at 12,000 gs. Discard flow through.
17.
Place spin column into microfuge tube.
Add 50uL of TE Buffer to center of column. Incubate column in room temp for 1
min.
18.
Centrifuge column at 12,000 rcfs for 2 mins. Discard column. Nanodrop
DNA. Store at 4C short term, -20C long term.
Nanodrop results-
A21:
A22:
A31:
A32:
B21:
B22:
B31:
B32:
None were sufficient enough to send for
sequencing. Likely errors also due to overgrowth of colonies.
PCR
Gradient PCR of only MC001 blocks
conducted.
Master mix of
75 uL created (calculate ratio of 1X x 7.5/5)
o Phusion 5X HF Buffer = 37.5 uL
o dNTPs 10 mM = 3.75 uL
o Phusion DNAP = 1.875 uL
o H2O =
116.25 uL
o DMSO = 5.625 uL
o DNA =3.75 uL
o F Primer MC003= 9.375 uL
o R Primer MC004 =9.375 uL
10uL Master mix aliquoted into 7
samples.
Thermocycler settings- 98o 2
min, 98o 15 s, 60o, 62o, 64o,
66o, 68o, 70o, 72o 30 s, 72o
1 min, 72o 10 min, 4o hold.
Actual Anneal temperatures- 60.0o,
62.0o, 63.3o, 66.6o, 68.2o, 69.7o,
72.0o
T=66.0o, G=6.0o
for 30 cycles
Gel Electrophoresis
1 % agarose gel run for 30 mins at
120V. 25 uL of samples loaded. 1kB plus Invitrogen
ladder loaded. Only sufficient product seen for block 2. Indicates issues with
block 1.
Image

8/25
Sequencing Results: Found a sample
(127) containing entire MC001 insert!
Innoculated sample 127 and prepared for induction with L-Rhamnose.
Re-plated TOPO reactions for MC002/3 as
overgrowth of colonies on previous plates.
Set up inductions however, cultured at
37C instead of 30C. Pay attention to this, this is important, all the protocols
have induced L-Rhamnose at 30C
8/26
Start over the inductions, follow the igem protocol file for help. You will need to make more LB.
I have already made a few glycerol stocks. You will need to set up a 2mL innoculation in a culture tube as well as a 40mL innoculation in a flask. The 40mL innoculation
will be used for inductions. [Finished]
Redo TOPO transformations.
8/27
8/28
8/31
Reviewed work done over past three
days. Discussed ways to resolve western blot issue. Seems to be a lot of
non-specific binding -we see blots of many different sizes. Blots of antibodied products are very bright relative to ladder.
Kevin mentioned degradation could be an issue (would cause lots of different
sizes in blots). On closer examination, 0% rhamnose
has no (barely any) protein. Updated notebook, e-mailed Justin and Kevin for
trouble shooting.
BH- Type up lysing assay and bradford assay -give some
specifics about western blot (how much each sample, and calculations)
E-mail sleep people
9/1
Induction Solutions
In order to re-do induction western
blots, made new 10% Rhamnose w/v solution by diluting
1 g of Rhamnose in 9 mL of water to bring final
volume to 10mL.
Culture
Set up two 40 mL cultures of sample 127
in 250 mL flasks. Added 20uL of Cam in each, 40 mL of LB and 10uL of 2 mL
sample 127 overnight culture. Note: 2mL overnight culture has been stored in
room temperature for past four days -may also have to start from new glycerol
stock. 40mL cultures placed in 37C incubator at 11:50.
9/2
Dilutions of Rhamnose
Inducer Samples
As discussed in meetings, errors with
previous western blot likely to be caused due to overexpression of pRhamnose. iGEM
team used low copy number plasmid. Re-made inducer solutions using 1/10 of %
from 0.001 - 0.1% in order to test lower gradient. Induction started at 2:30 am
when OD600 of 40 mL innoculations was 1.6(?)
Minimal Media
Ordered M9 minimal media 5X salts on
Rust Lab tab. Should take till friday
to arrive.
Restriction Digest
Protocol from iGEM.
Digest
§ Enzyme Master Mix for Plasmid Backbone (25ul
total, for 5 rxns)
§ 5 ul NEB Buffer 2
(use CutSmart)
§ 0.5 ul BSA
§ 0.5 ul EcoRI-HF
§ 0.5 ul PstI
§ 0.5 ul DpnI (Used to digest any template DNA from
production)
§ 18 ul dH20
§ Digest Plasmid Backbone
§ Add 4 ul linearized
plasmid backbone (25ng/ul for 100ng total)
§ Add 4 ul of Enzyme
Master Mix
§ Digest 37C/30 min, heat kill 80C/20 min
Ligation
§ Add 2ul of digested plasmid backbone (25 ng)
§ Add 1 ul T4 DNA ligase buffer. Note: Do not use quick ligase
§ Add 0.5 ul T4 DNA ligase
§ Add water to 10 ul
§ Ligate 16C/30 min, heat kill 80C/20 min
§ Transform with 1-2 ul
of product
Restreaked TOPO plate
Plates of successful TOPO reactions for
MC002_b1 and MC003_b1. The colonies on these plates had overgrown -so in an
attempt to isolate one colony for miniprepping, these
colonies were streaked onto new Cam plates.
Transformed new TOPO reactions for
MC02/3 and BH002/3
New TOPO reactions were conducted in
parallel with the streaking.
Set up Gibson of BH002/3
BH002 and BH003 were gibsonsed with their respective Amp backbones and Cam backbones.
9/3
Lysed cells after induction
Cells were lysed after 10 hours of
induction. Cells resuspened in lysis buffer,
transferred to microfuge tube with 0.1 mL glass beads, vortex for 30 s and incuabted on ice for 1 min (repeated 5 times).
Bradford Assay
Bradford assay conducted to quanity amount of protein added. 1 mL 1x bio-rad protein reagent and 0.2 uL
of protein sample used for each spectrophotometer (595 nm) reading. BSA protein
standards with fixed concentrations made and absorbance measured for generating
standard curve (concentration vs absorbance).
Conducted Western blot
SDS-Page gel
SDS-Page gel set up with purified
proteins, cyanobacterial lysate and ecoli vector only
lysate (not used in this specific assay). Proteins purified and loaded on
14-20% gel in 2 fold diluations from 100 ng to 1.56
ng. Proteins loaded with 3 x loading dye and denatured at 70C for 10 mins.
Empty lanes loaded with 1 x loading dye. Gel run at 300 v for 22 mins.
Transfer
Transfer constructed with membrane
sandwich -plastic latched case, two whatman papers,
two sponges, membrane, gel. Transfer apparatus run for
1.5 hours at 90 V.
Blocking and Staining
Memabrane blocked with 2% dry milk and TBST for 1 hour. Membrane pieces
stained with primary Kai rabbit antibodies for 1.5 hours. Membrane washed with
washing buffer for 10 mins for 3 times (30 mins total). Membrane stained with
secondary goat antibody for 1 hour. Membrane peices
washed with washing buffer for 10 mins for 3 times (30 mins total). Membrane
imaged.
Ran colony PCR of TOPO reactions
Only MC003 TOPO worked. BH002/3 Gibsons worked, no TOPO results for
MC002, BH002, BH003.
Inoculated negative control
Ecoli with vector (Cam backbone) innoculated
at 37C. 2 mL innoculation generated.
Image

KaiB and KaiA gave no clear results.
9/4/15
Set up Rhamnose
time course experiment and rhamnose gradient
Rhamnose time course experiment conducted in both M9 supplemented media
(4% glycerol, 0.1% casamino acids, antibiotic) and
LB. 40 mL culture grown overnight. Cells pelleted, washed with 1 mL water,
pelleted then reintroduced into Lb or M9 media. Time
course consisted of induction at 30C for 10 hours. 0.5% Rhamnose
used. Time course experiment conducted in order to examine behavior of KaiA throughout induction. 2 mL of cells frozen every 2
hours for 16 hours. Shocks also conducted for 1 hour and 6 hours. Shocks
conducted to emulate synchronization method -incubation in M9 media with no
supplements to limit ATP available to cells. Samples after shock collected at 1
hour, 6 hours, and 62 hours.
Rhamnose gradient assay also conducted, using smaller gradient with 10
fold dilution. Induction conducted for 10 hours.
9/7/15
MC003, BH002, and BH003 colonies were
submitted for sequencing in UChicago sequencing
facility with respective primers. MC003 used specific primers, BH002/3 used M13
and M14 primers as they were in TOPO vector.
9/8/15
Bradford Assay
Bradford assay conducted to quanity amount of protein added. 1 mL 1x bio-rad protein reagent and 0.2 uL
of protein sample used for each spectrophotometer (595 nm) reading. BSA protein
standards with fixed concentrations made and absorbance measured for generating
standard curve (concentration vs absorbance).
Conducted Western blot
SDS-Page gel set up with purified
proteins, cyanobacterial lysate and ecoli vector only
lysate. Proteins purified and loaded on 14-20% gel in 2 fold diluations from 100 ng to 1.56 ng. Proteins loaded with 3 x
loading dye and denatured at 70C for 10 mins. Empty lanes loaded with 1 x
loading dye. Gel run at 300 v for 22 mins. Kaleidoscope ladder used.
Transfer constructed with membrane
sandwich -plastic latched case, two whatman papers,
two sponges, membrane, gel. Transfer apparatus run for
1.5 hours at 90 V.
SDS-Page gel
SDS-Page gel set up with purified
proteins, cyanobacterial lysate and ecoli vector only
lysate (not used in this specific assay). Proteins purified and loaded on
14-20% gel in 2 fold diluations from 100 ng to 1.56
ng. Proteins loaded with 3 x loading dye and denatured at 70C for 10 mins.
Empty lanes loaded with 1 x loading dye. Gel run at 300 v for 22 mins.
9/9
Blocking and Staining
Membrane blocked with 2% dry milk and
TBST for 1 hour. Membrane pieces stained with primary Kai rabbit antibodies for
1.5 hours. Membrane washed with washing buffer for 10 mins for 3 times (30 mins
total). Membrane stained with secondary goat antibody for 1 hour. Membrane
pieces washed with washing buffer for 10 mins for 3 times (30 mins total).
Membrane imaged. Blot only conducted for KaiA and KaiC as issues with obtaining KaiB
antibodies.
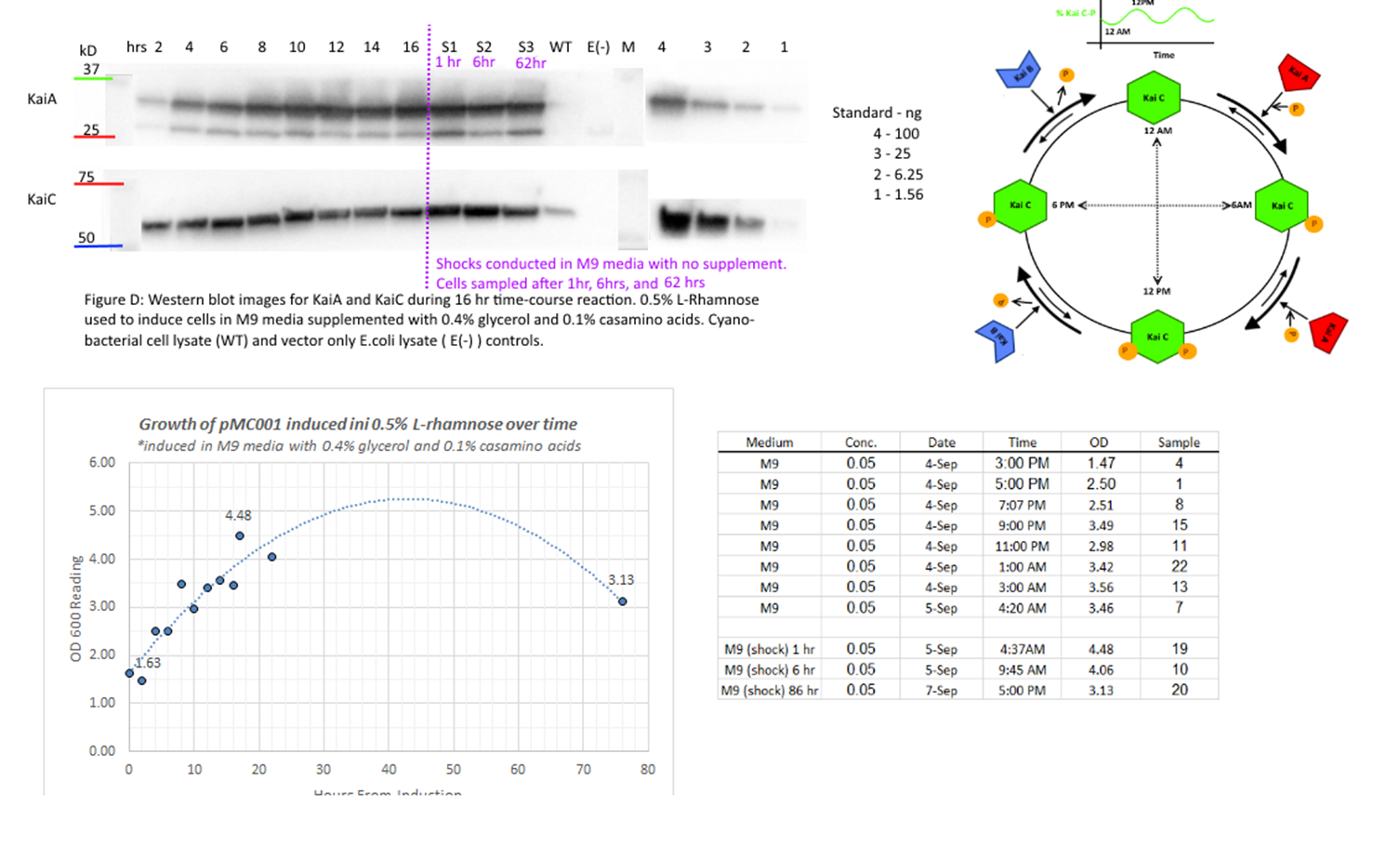
9/10
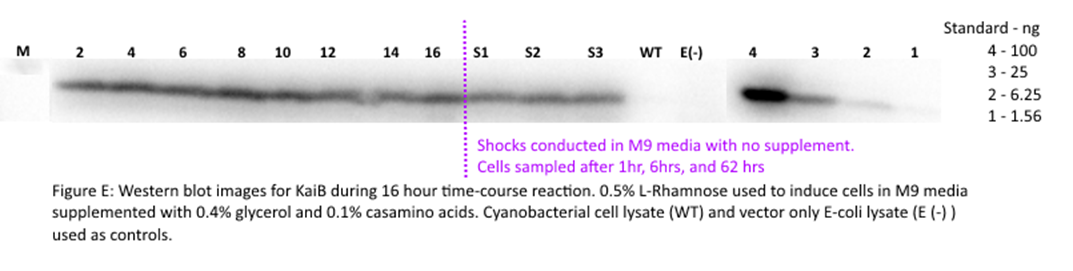
Induction of Rhamnose
with lower gradient started again. Finished western blot for Kai B from
previous assay. Ran PCR of block MC002_b1, MC003_b1. Gel extracted blocks.

9/11
Bradford Assay
Bradford assay conducted to quanity amount of protein added. 1 mL 1x bio-rad protein reagent and 0.2 uL
of protein sample used for each spectrophotometer (595 nm) reading. BSA protein
standards with fixed concentrations made and absorbance measured for generating
standard curve (concentration vs absorbance).
Conducted Western blot
SDS-Page gel
SDS-Page gel set up with purified
proteins, cyanobacterial lysate and ecoli vector only
lysate (not used in this specific assay). Proteins purified and loaded on
14-20% gel in 2 fold diluations from 100 ng to 1.56
ng. Proteins loaded with 3 x loading dye and denatured at 70C for 10 mins.
Empty lanes loaded with 1 x loading dye. Gel run at 300 v for 22 mins.
Transfer
Transfer constructed with membrane
sandwich -plastic latched case, two whatman papers,
two sponges, membrane, gel. Transfer apparatus run for
1.5 hours at 90 V.
Ran another PCR of MC002_b1, MC003_b1
Ran PCR of gel extracted blocks
MC002_b1 and MC003_b1
9/12
Blocking and Staining
Ponceau staining conducted -membranes incubated in ponceau
reagent for 5 mins. Could see faint bands for KaiA
and KaiC samples. Membrane pieces blocked with 2% milkd and TBST overnight.
9/13
No primary antibody available at time,
continued to block.
9/14
Membrane pieces stained with primary
Kai rabbit antibodies for 1.5 hours. Membrane washed with washing buffer for 10
mins for 3 times (30 mins total). Membrane stained with secondary goat antibody
for 1 hour. Membrane pieces washed with washing buffer for 10 mins for 3 times
(30 mins total). Membrane imaged. Only kai B showed up in image.
9/15
Induction
Induction repeated. Cells pelleted,
washed with 1 mL water, pelleted then reintroduced into Lb
or M9 media. Concentrations:
Like our team Facebook page, Genehackers@UChicago!
Questions? Comments? Send us an email!