Difference between revisions of "Team:KU Leuven/Modeling/Internal"
| Line 145: | Line 145: | ||
</p> | </p> | ||
| − | <div class="datatable"> | + | <div class="datatable" style="width:90%"> |
<table> | <table> | ||
<tr> | <tr> | ||
| Line 202: | Line 202: | ||
<p> | <p> | ||
The results are in table 1. The program gives us results in au (arbitrary units). Since we know the translation rate of LuxI and LuxR, we can use these as a base to calculate the other translation rates since the used scale is proportional. (table 1, column 2) LuxI and LuxR have almost the same output from the calculator which corresponds with the paper where they also have the same translation rate. (Ag43 has a very low translation rate, which is not completely illogical, since Ag43 is by far the biggest protein.) Our values are normal values since 1000 is a moderate value and values between 1 and 100 000 are possible. (efficient search, mapping and optimization, salis). We did get warnings about the prediction (NEQ: not at equilibrium) which happens when mRNA may not fold quickly to its equilibrium state. </p> | The results are in table 1. The program gives us results in au (arbitrary units). Since we know the translation rate of LuxI and LuxR, we can use these as a base to calculate the other translation rates since the used scale is proportional. (table 1, column 2) LuxI and LuxR have almost the same output from the calculator which corresponds with the paper where they also have the same translation rate. (Ag43 has a very low translation rate, which is not completely illogical, since Ag43 is by far the biggest protein.) Our values are normal values since 1000 is a moderate value and values between 1 and 100 000 are possible. (efficient search, mapping and optimization, salis). We did get warnings about the prediction (NEQ: not at equilibrium) which happens when mRNA may not fold quickly to its equilibrium state. </p> | ||
| − | <div class="datatable"> | + | <div class="datatable" style="width:90%"> |
<table> | <table> | ||
<tr> | <tr> | ||
| Line 263: | Line 263: | ||
Before the proteins can bind the promoter region, they first have to make complexes. cI and penI form homodimers, while LuxR first forms a heterodimer with AHL and afterwards forms a homodimer with another LuxR/AHL dimer. This next part will describe the kinetics of such complexation. We only need the parameters of LuxR and cI, because the Hill function found for penI already was adapted for the protein in monomer form. <br> | Before the proteins can bind the promoter region, they first have to make complexes. cI and penI form homodimers, while LuxR first forms a heterodimer with AHL and afterwards forms a homodimer with another LuxR/AHL dimer. This next part will describe the kinetics of such complexation. We only need the parameters of LuxR and cI, because the Hill function found for penI already was adapted for the protein in monomer form. <br> | ||
<br></p> | <br></p> | ||
| − | <div class="datatable"> | + | <div class="datatable" style="width:90%"> |
<table> | <table> | ||
<tr> | <tr> | ||
| Line 336: | Line 336: | ||
<br> | <br> | ||
<p>Yang et al. (2005) studied these reactions and found a method to estimate the constants. Their constants of this reversible Ping-Pong Bi-Bi reactions are put in the next table : </p> | <p>Yang et al. (2005) studied these reactions and found a method to estimate the constants. Their constants of this reversible Ping-Pong Bi-Bi reactions are put in the next table : </p> | ||
| − | <div class="datatable"> | + | <div class="datatable" style="width:90%"> |
<table> | <table> | ||
<tr> | <tr> | ||
| Line 403: | Line 403: | ||
<p>Proteins, mRNA and other metabolites have a turnover rate. They are degraded over time. The degradation rate will be described as proportional to the amount of biomolecules. The coefficient of proportionality d is the degradation constant. Not every molecule has the same degradation rate, since some molecules are more stable than others. We can influence the stability of the molecules. For example, in cell B it is important that there is a fast switch between conditions and a fast turnover of CheZ and RFP is necessary. This is why we add a LVA-tag to these proteins. This tag destabilizes the protein and makes them degradade faster. For TransaminaseB we choose a very high degradation rate, because we did not include degradation terms for the TransaminaseB bound to substrate. The degradation rates used in the model are put in the next table: <p> | <p>Proteins, mRNA and other metabolites have a turnover rate. They are degraded over time. The degradation rate will be described as proportional to the amount of biomolecules. The coefficient of proportionality d is the degradation constant. Not every molecule has the same degradation rate, since some molecules are more stable than others. We can influence the stability of the molecules. For example, in cell B it is important that there is a fast switch between conditions and a fast turnover of CheZ and RFP is necessary. This is why we add a LVA-tag to these proteins. This tag destabilizes the protein and makes them degradade faster. For TransaminaseB we choose a very high degradation rate, because we did not include degradation terms for the TransaminaseB bound to substrate. The degradation rates used in the model are put in the next table: <p> | ||
| − | <div class="datatable"> | + | <div class="datatable" style="width:90%"> |
<table> | <table> | ||
<tr> | <tr> | ||
| Line 531: | Line 531: | ||
With this approximation we get more logical results. We take the diffusion rate for AHL equal to 0.23 1/s. For Leucine we make a differce between inward and outward diffusion. Inward diffusion is facilitated by transporters, so we choose this value to be the largest. The inward diffusion rate is </p> | With this approximation we get more logical results. We take the diffusion rate for AHL equal to 0.23 1/s. For Leucine we make a differce between inward and outward diffusion. Inward diffusion is facilitated by transporters, so we choose this value to be the largest. The inward diffusion rate is </p> | ||
| − | <div class="datatable"> | + | <div class="datatable" style="width:90%"> |
<table> | <table> | ||
<tr> | <tr> | ||
Revision as of 08:59, 17 September 2015

Internal Model
1. Introduction
We can think of many relevant questions in implementing a new circuit: how sensitive is the system, how much will it produce and will it affect the growth? As such, it is important to model the effect of the new circuits on the bacteria. This will be done in the Internal Model. We will use two approaches. First we will use a bottom-up approach. This involves building a detailed kinetic model with rate laws. We will use Simbiology and ODE's to study the sensitivity and dynamic processes inside the cell. This is the bottom-up approach. Afterwards, a top-down model, Flux Balance Analysis (FBA), will be used to study the steady-state values for production flux and growth rate. This part is executed by the iGEM Team of Toulouse as part of a collaboration and can be found here
2. Simbiology and ODE
In the next section we will describe our Simbiology model. Simbiology allows us to calculate systems of ODE's and to visualize the system in a diagram. It also has options to make scans for different parameters, which allows us to study the effect of the specified parameter. We will focus on the production of leucine, Ag43 and AHL in cell A and the changing behavior of cell B due to changing AHL concentration. In this perspective, we will make two models in Simbiology: one for cell A and cell B. First we will describe how we made the model and searched for the parameters. Afterwards we check the robustness of the model with a parameter analysis and we do scans to check for the effects of molecular noise.
3. Quest for parameters
We can divide the different processes that are being executed in the cells in 7 classes: transcription, translation, DNA binding, complexation and dimerization, protein production kinetics, degradation and diffusion. We went on to search the necessary parameters and descriptions for each of these categories. To start making our model we have to chose a unit. We choose to use molecules as unit because many constants are expressed in this unit and it allows us to drop the dillution terms connected to cell growth. We will also work with a deterministic model instead of a stochastic model. A stochastic model will show us the molecular noise, but we will check this with parameter scans.
The next step is to make some assumptions:
- The effects of cell division can be neglected
- The substrate pool can not be depleted and the concentration (or amount of molecules) of substrate in the cell is constant
- The exterior of the cell contains no leucine at t=0 and is perfectly mixed
- Diffusion happens independent of cell movement and has a constant rate
4. System
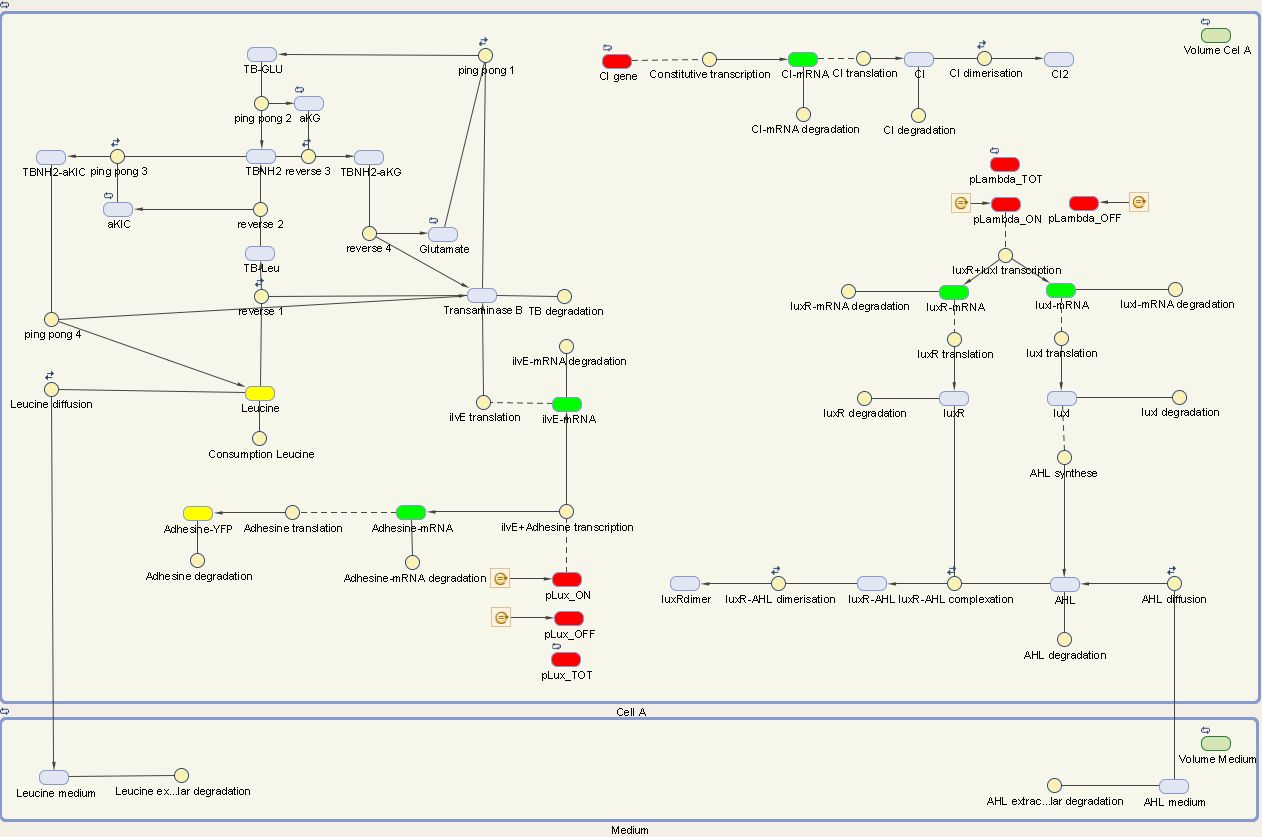
The designed circuit in Cell A is under control of a temperature sensitive cI repressor. Upon raising the temperature, cI will dissociate from the promoter and the circuit is activated. This leads to the initiation of the production of LuxR and LuxI. LuxI will consecutively produce AHL, which binds with LuxR. The newly formed complex will then activate the production of Leucine and Ag43. Leucine and AHL are also able to diffuse out of the cell into the medium. Ag43 is the adhesine which aids the aggregation of cells A, while Leucine and AHL are necessary to repel cells B.
We can extract the following ODE's from this circuit:
Cell A equations
Symbols:${}$ ${\alpha}$: transcription term, ${\beta}$: translation term, $d$: degradation term,
$D$: diffusion term, ${ K_d}$: dissociation constant, n: Hill coefficient, L: leak term
$$\frac{{\large d} m_{cI}}{d t} = \alpha_1 {\cdot} cI_{gene} - d_{mCI} {\cdot} m_{cI}$$ \begin{align} \frac{{\large d}{cI}}{d t} = \beta_{cI} {\cdot} {m_{cI}} -2 {\cdot} {k_{cI,dim}} {\cdot} {cI}^2 + 2 {\cdot} {k_{-cI,dim}}{\cdot} {[cI]_2} - d_{cI} {\cdot} {cI} \end{align}
We visualize these ODE's in the Simbiology Toolbox which results in the following diagram:
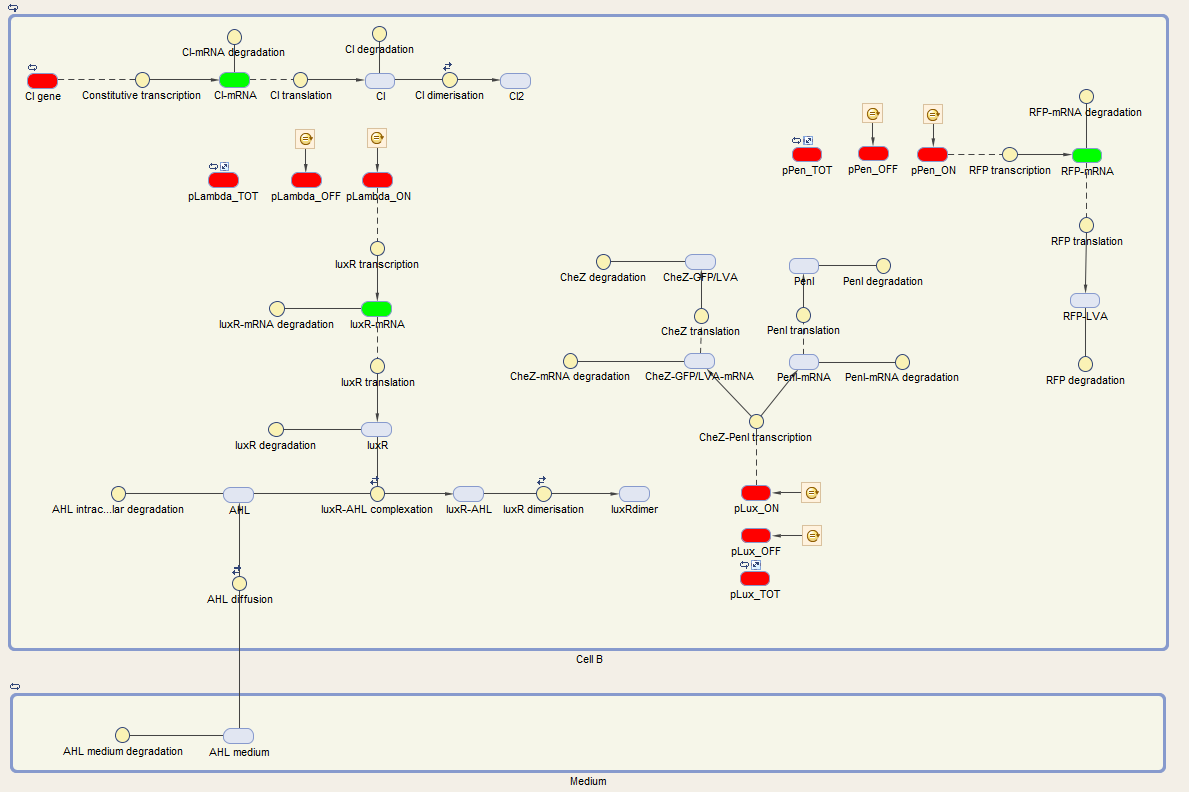
The system of Cell B is also under control of the cI repressor and is activated similar as cell A. The activation by the temperature raise, leads to the production of LuxR. AHL of the medium can diffuse into the cell, binding LuxR and activating the next component of the circuit. This leads to the production of CheZ and PenI. CheZ is the protein responsible for cells to make a directed movement, governed by the repellent Leucine. PenI is a repressor which will shut down the last part of the circuit which was responsible for the production of RFP.
We can extract the following ODEs for Cell B from this sytem:
Cell B equations
Symbols: ${\alpha}$:transcription term, ${\beta}$:translation term, $d$:degradation term,
$D$:diffusion term, ${ K_d}$:dissociation constant, n:Hill coefficient, L:leak term
$$\frac{{\large d} m_{cI}}{d t} = \alpha_1 {\cdot} cI_{gene} - d_1 {\cdot} m_{cI}$$ $$\frac{{\large d}{cI}}{d t} = \beta_1 {\cdot} {cI} -2 {\cdot} {k_{cI,dim}} {\cdot} {cI}^2 + 2 {\cdot}{k_{-cI,dim}}{\cdot} {[cI]_2} - d_{cI} {\cdot} {cI} $$
We visualize these ODE's in the Simbiology toolbox. This gives us the following diagrams:
5. Results
Cell A graph of all, graph of Leucine, graph of AHL
Cell B graph of all, graph with induction and without induction
Sensitivity analysis
Conclusion and discussion








