Team:Waterloo/Modeling/PAM Flexibility
Modeling Engineered Cas9 PAM Flexibility
CRISPR-Cas9 is one of the most significant advances in genetic engineering in the last several years. There are multiple examples in nature of a CRISPR-Cas9 system being used for as a defense against infection. All of these use their own variant of the Cas9 protein to perform nuclease activity in a region dependent on the presence and location of a system-specific protospacer adjacent motif (PAM) sequence. From these, it has been observed that naturally occurring PAM sequences can be quite different from each other. For example, S. pyogenes uses a PAM sequence of NGG while S. thermophilus has a PAM sequence of NAAGAW in its CRISPR1-Cas system and NGGNG in its CRISPR3-Cas system. In current genetic engineering technologies, it is mostly the S. pyogenes CRISPR-Cas9 (spCas9) protein which is used.
However, one limitation of spCas9 is the NGG PAM required for binding to the target DNA site. While the 20bp protospacer can be easily specified with different sgRNAs, the PAM is essentially hardcoded into the Cas9 protein structure of the PAM-interacting (PI) domain. This limits the number of possible targets in a given genome, and thus the possible uses of spCas9 for genetic engineering. However, attempts to selectively modify Cas9 to bind new PAMs, specifically by changing two amino acids thought to bind directly to the PAM DNA sequence, failed.
Kleinstiver et al. recently demonstrated modified spCas9 with altered PAM specificity. Cells were transformed with a plasmid bearing a toxic gene and an NGA adjacent protospacer, as well as a spCas9 gene with a randomly mutagenized PAM Identification Domain. Only cells capable of binding the NGA PAM sequence were able to cut the plasmid, and thus survive. By sequencing surviving clones, the authors were able to identify mutations in the PI domain that altered PAM sequence specificity.
Two mutants in particular showed both high specificity and activity for the NGA PAM sequence. The first mutant, referred to as VQR (D1135V,R1335Q,T1337R), worked equivalently on all four NGAN PAM sequences. The second mutant, EQR (D1135E, R1335Q, T1337R) was most specific to NGAG PAMs. A second round of testing also found a VRER variant ((D1135V,G1218R,R1335E,T1337R) that was specific to NGCG PAMs.

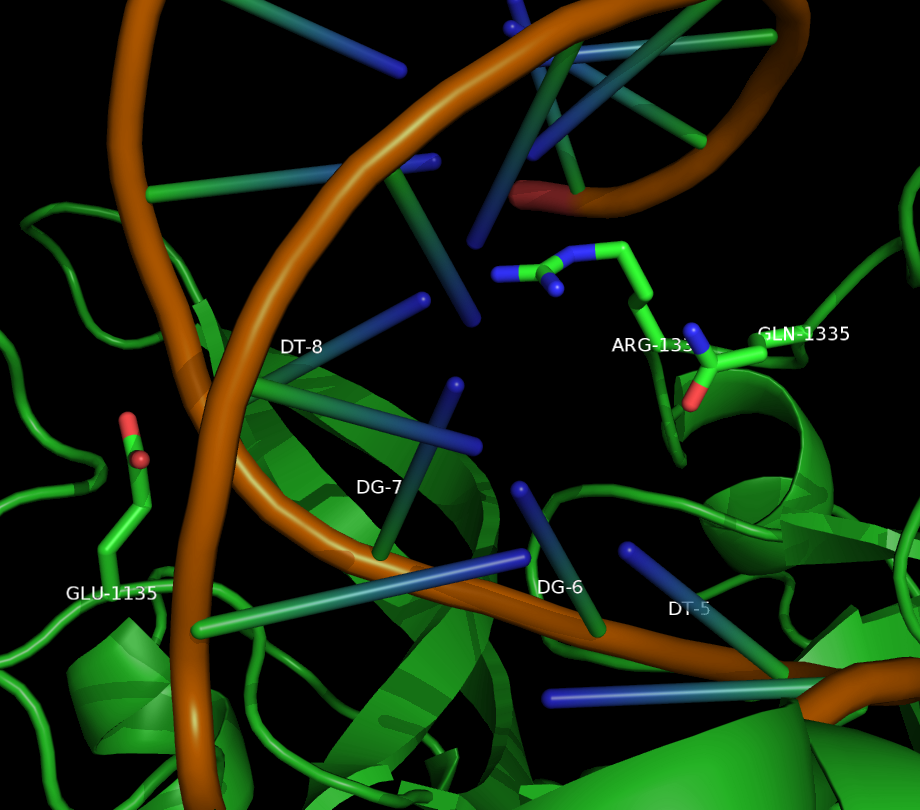
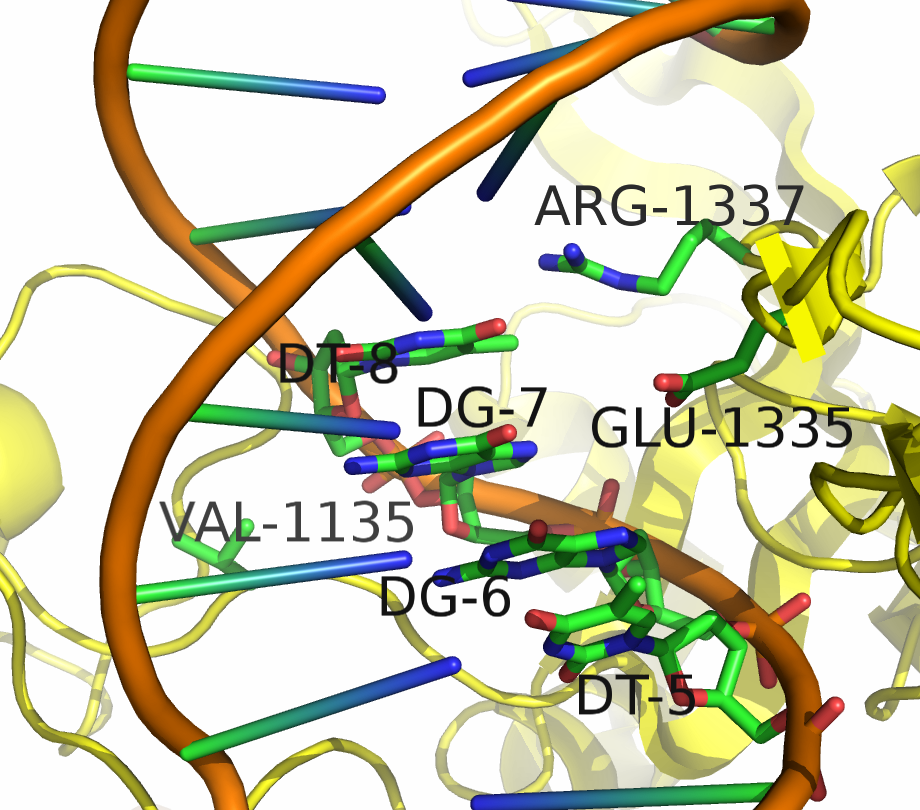
These result indicate that spCas9 can display altered PAM specificity with relatively few mutants. Additionally, the data produced by Kleinstiver et al. also provided significant insight into the types of mutations that altered PAM specificity. This allows for a more targeted approach to engineering new spCas9 PAMs.
Computational Engineering of PAM Flexibility
The goal our work was developing a procedural pipeline to produce and identify Cas9 mutants with altered PAM specificity. This would be done by performing simulations of spCas9 mutants docking to a PAM DNA sequence to predict a theoretical binding affinity for many different PAM variants. Finally, the specificity and affinity of each spCas9 mutant would be determined by clustering in order to find mutants which will be highly specific for particular PAM sequences, which will then be tested in the lab.

Obtaining a Structural Model of spCas9
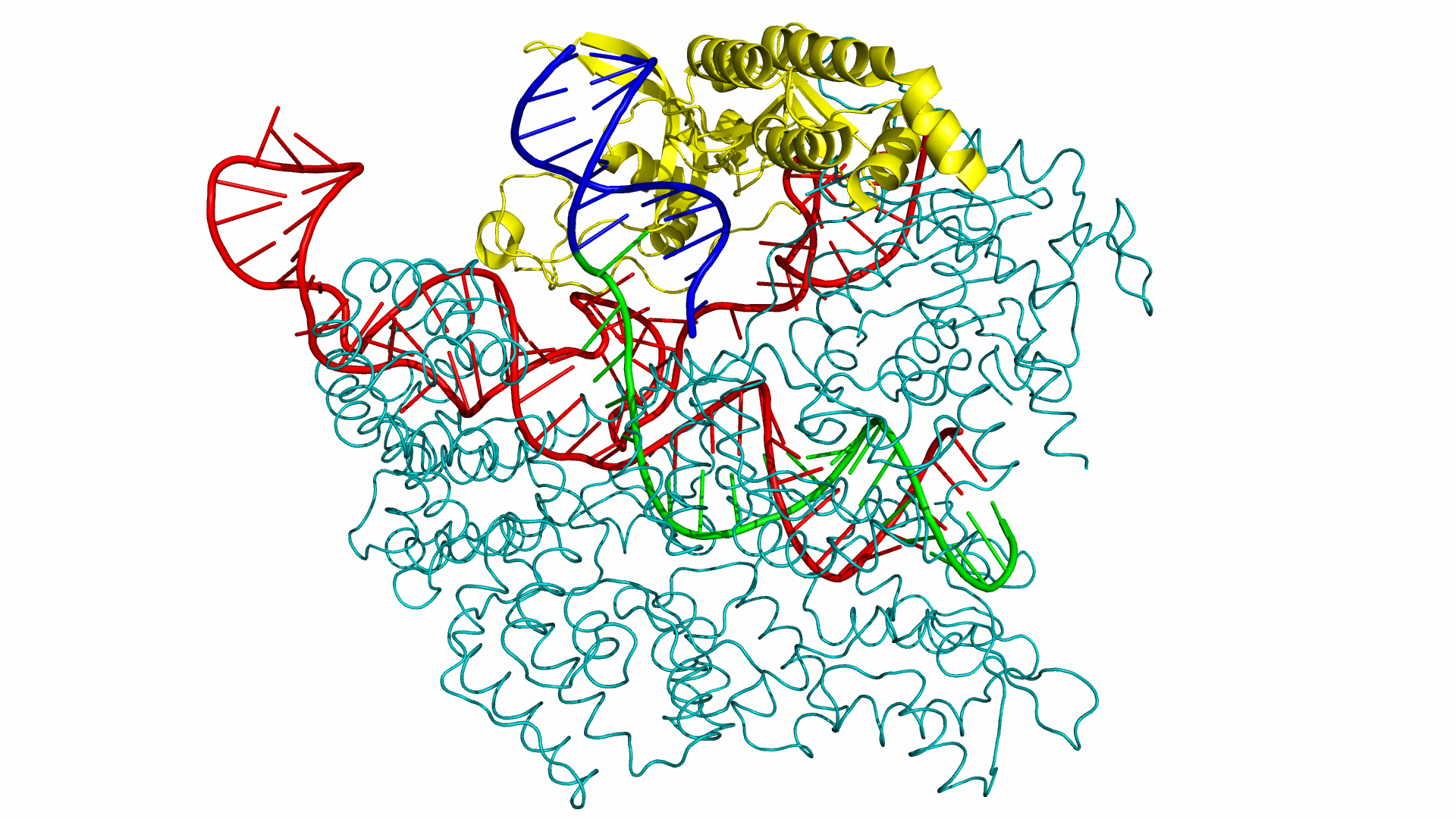
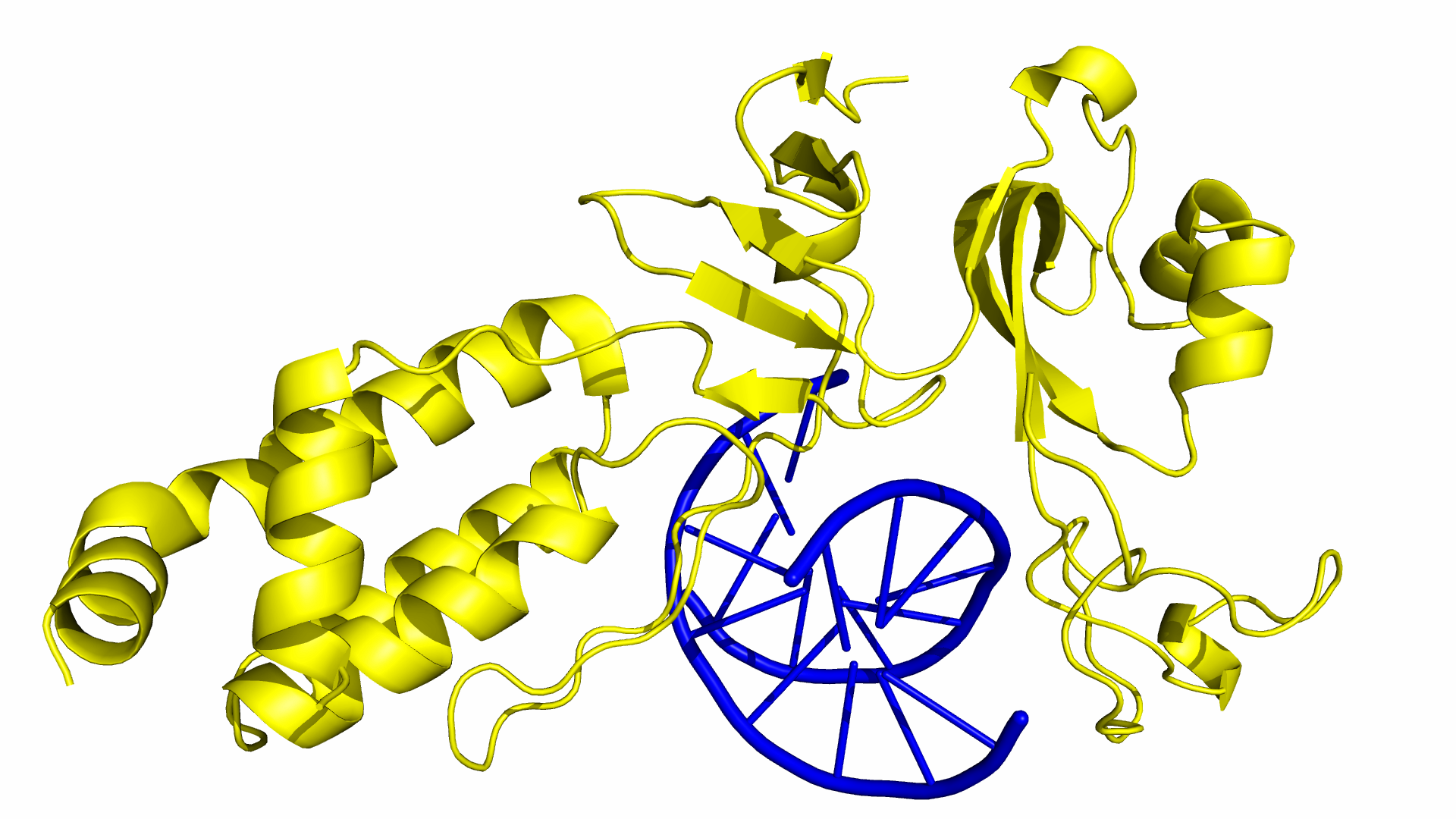
As of June 2015, at least three different groups have produced experimental data on Cas9 structure. In particular, the PDB we used was produced by Anders et al. By using a catalytically dead version of spCas9 (dCas9), Anders et al. managed to determine the crystal structure of a dCas9 protein bound to both the sgRNA and the double stranded target DNA. This data was analyzed to mechanism by which spCas9 binds to the PAM site and the target DNA. The PDB file containing this structure can be found in the Protein Data Bank with ID 4UN3.
Creating Computational Models of spCas9 Mutants
We developed a script that allows for mutations in the region of interest of the Cas9 protein that interacts with the PAM sequence in DNA. Our script induced in silico mutations resulting in a change of amino acid sequence in the Cas9 protein. The type of amino acid substitutions produced by our script were position-specific; this constraint was based on data from Kleinstiver et al. in order to restrict our sample space to mutants that were likely to produce mutants with an affinity to some PAM sequence. Additional constraints were imposed based on known biochemical properties of the amino acids being mutated.
Once a mutant has been generated, a full set of pdbs with the 64 or 256 different PAM sequence variants is created to test affinity for all possible PAM sites.
Our approach for determining the PAM sequence affinity of each Cas9 mutant is by generating a combination of either 64 or 256 different PAM sequences using both 3DNA and Chimera (our choice of software will be elaborated below). These combinations were implemented by altering the original 4UN3 PDB file and were subsequently tested and scored using PyRosetta and visualized through PyMOL.
Modeling Docking with PyMOL and PyRosetta
Simulation of Cas9 interactions with the PAM region were performed with PyRosetta. Docking was performed between wildtype/mutant Cas9 and all PAM variants, in order to determine the relative stability of the Cas9 candidate and PAM sequence. Structures that result in a low docking score (and thus high stability) indicate that this complex is predicted to occur in real-life, and that this Cas9 mutant should bind and cut sequences with that PAM sequence. PyMOL was used to observe the docking and repacking protocols and ensure that appropriate actions were being done in our simulation runs.
Analysis of Simulation Data
The docking score of these simulations is tracked over multiple runs. The scores are then averaged and mutant specificity to different PAM sequences is determined, based on the clustering of the scores by PAM. Mutants highly specific for a particular PAM will be tested in the lab as they are good candidates for Cas9 mutants with altered PAM specificity.
Engineering Pipeline
Combining these steps together, the overall approach to identifying possible mutants for new PAM variants is:
- Start with the pdb with crystal structure data for spCas9 bound to the target DNA
- Modify the amino acid sequence of the PDB. Mutants will be generate based on the location and amino acid substitution data from Kleinstiver et al., as well as known biochemical properties of the amino acid being mutated.
- Once a mutant has been generated, a full set of pdbs with the 64 or 256 different PAM variants will be created for each mutant.
- The DNA sequence is docked to the PI domain using PyRosetta, with scores being tracked over multiple runs.
- Scores are averaged and mutant specificity to different PAM sequences is determined, based on the clustering of the scores by PAM.
- Mutants highly specific for a particular PAM will be tested in the lab.
Model Validation
The first test of our proposed pipeline was to determine if we could reproduce the PAM specificity of wild type spCas9 in silico. The pdb file was run against all 64 possible 3 nucleotide pams. The results of the initial scoring can be seen below.
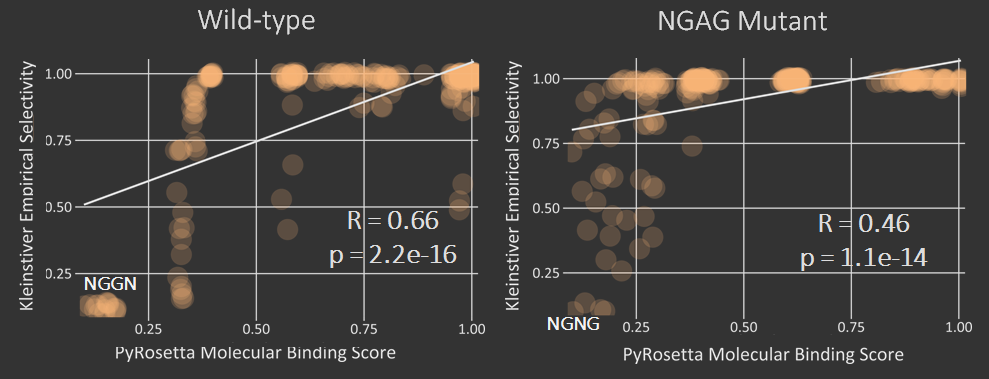
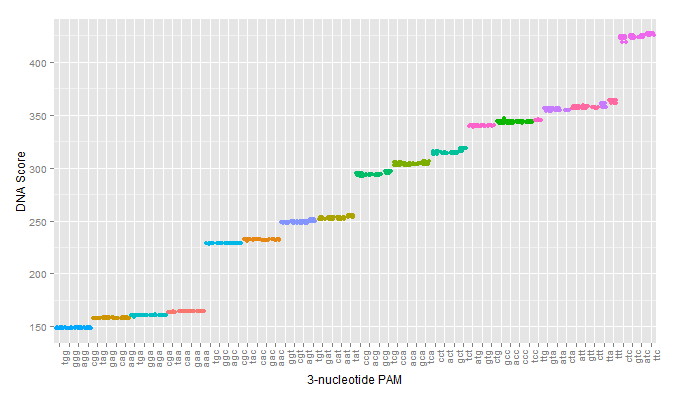
A modification that was made to our intial procedure was to reduce the size of the Cas9-sgRNA-DNA model. Instead of including the entire Cas9 structure, the PDB file was modified to include only the region of the Cas9 structure where interaction between Cas9 and the PAM site occurred. This caused each docking simulation to take less than 100 seconds to run, as opposed to over 1000 seconds with the full model, allowing us to greatly increase our throughput in determining Cas9 mutants of interest.
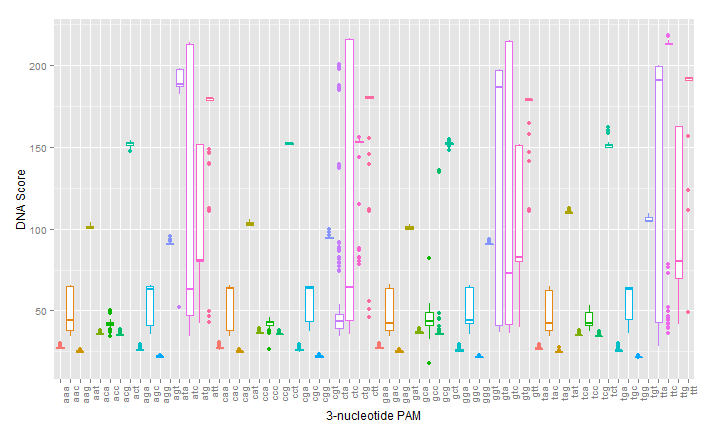
As can be seen in the figures, the trimmed Cas9 pdb performs just as well at predicting dockings, while signficantly reducing run time.
Mutant Cas9 PAM Affinities show Mixed Results
The next step in developing the tool was recreating mutants with known PAM specificity. The VQR and EQR mutants developed by Kleinstiver et al. were obvious choices for verify in silico modeling. However, the docking produced mixed results. While the expected NGA/NGAG PAMs did rank highly for binding affinity, the simulation incorrectly identified NGG and NAG PAMs as binding with equal affinity.
However, a simulation of the R1333Q/R1335Q mutant attempted by Anders et al. failed to distinguish between different PAM site, just as the experiment did.
Framework and Future Work
Proposed Tool
The software used for this project includes: PyRosetta used for protein design and analysis, PyMOL for protein visualization, as well as 3DNA and UCSF Chimera for structural analysis.
Our developed guide to PyRosetta and PyMOL on Linux, Mac OSX and Windows can be found on the documentation of our github wiki.
Future Work
Naturally, the next step in the progression of the project would be to rationally design a novel Cas9 protein with respect to the generated non-canonical PAM sequences, and to test the binding of the novel Cas9 DNA complex in order to achieve a completely in silico designed protein that has the ability to recognize a novel PAM sequence.
For this project, the interaction between Cas9 and the PAM region was ensured by further specifying physical fields such as the angle of torsion, degrees of freedom with respect to movement of the ligands, and repacking which allowed for the optimal conformation of the newly mutated protein so that conditions were ideal for binding. We hope to eventually include the Gibb's Free Energy value obtained from the interaction, as well as additional fields imposed mutation protocol by using other methods available such as the usage of a Task Factory and improving on the stringency of our current mutation script that implements the built-in mutate base function.
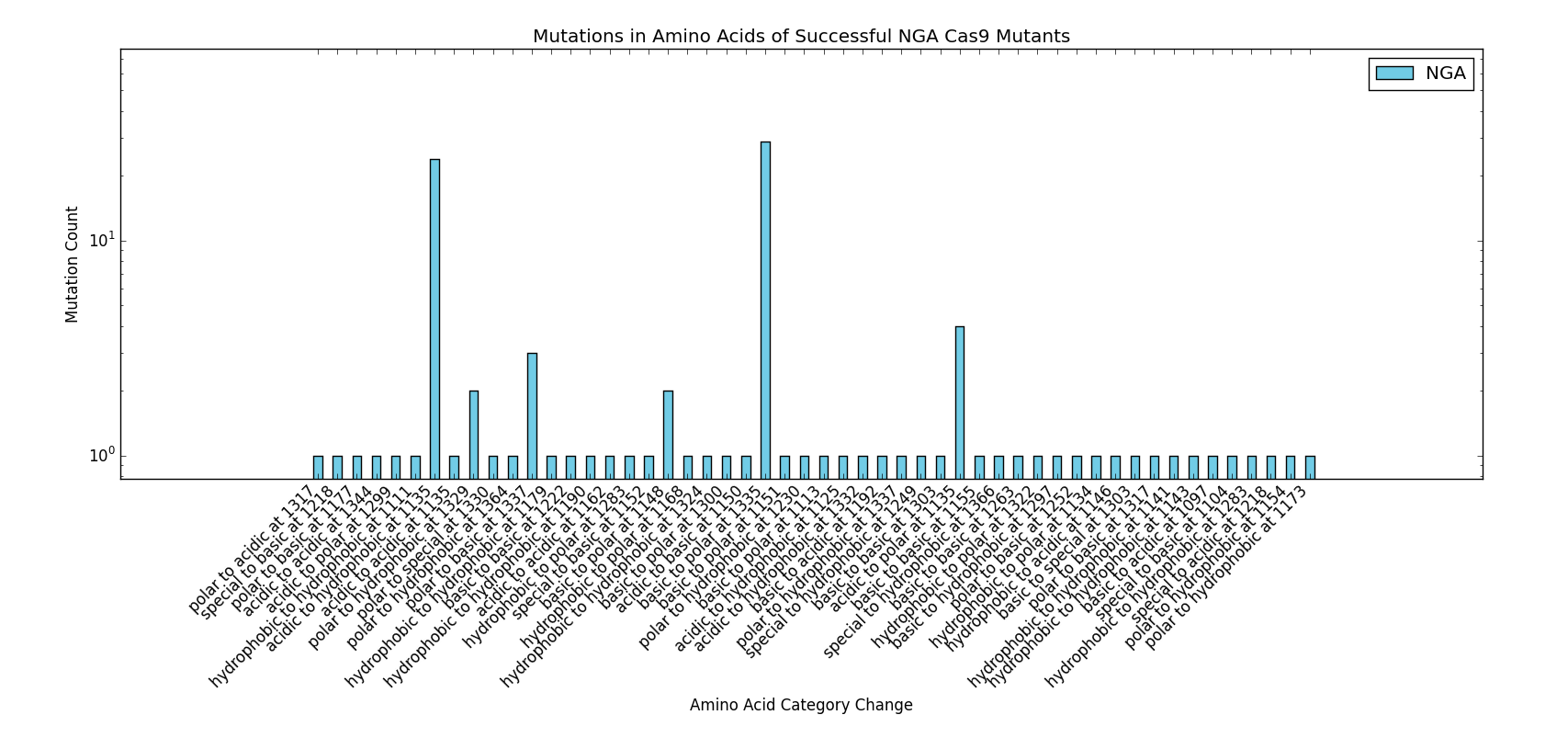
This particular area of the project used a significant amount of data published by the Kleinstiver et al. paper to perform the analyses and protocols presented. We would like to thank the authors of the "Engineered CRISPR-Cas9 nucleases with altered PAM specificities" paper for providing the requested figures and data used for the design of this section of the project. Future work done in the area of rational Cas9 design would benefit from initial analysis of the methods and data provided by Kleinstiver et al.