Team:TJU/Protocol


1 Parts Construction
July 15th, 2015.
Constructed J23100-ldhE-B0014, and linked them with pSB1C3.


Figure: ldhE
July 23th, 2015
Constructed Sequence 1.


Figure: Sequence 1.
August 7th, 2015
Synthesized J23103/J23100-rcfB -B0032/B0034 and linked them with pSB1C3.


Figure: rcfB-pSB1C3
Synthesized mf-lon-T7 terminator (Sequence 2).


Figure: mf-lon-T7 terminator
August 17th, 2015
Synthesized P170 using SOE-PCR protocol.

August 18th, 2015
Constructed J23100-RFP-B0014.


figure: RFP-B0014-pSB1C3
August 19th, 2015
Restriction enzyme digestion RFP and P170. And linked these part to pSB1C3.


August 27th, 2015
Knocked out gene pfl in E.coli MG1655.

Figure: pfl knock out.
September 1st, 2015
Construct RFP-TAG and linked it with pSB1C3.


Figure: RFP-TAG-pSB1C3.
2 MFC
July 30th, 2015
Experiment 1: Exploring suitable carbon source for Shewanella.
We constructed 3 groups of MFCs which had Shewanella + lactate + riboflavin, Shewanella + lactate, Shewanella + glucose respectively.

August 9th, 2015
Experiment 2: Exploring bacteria consortia.
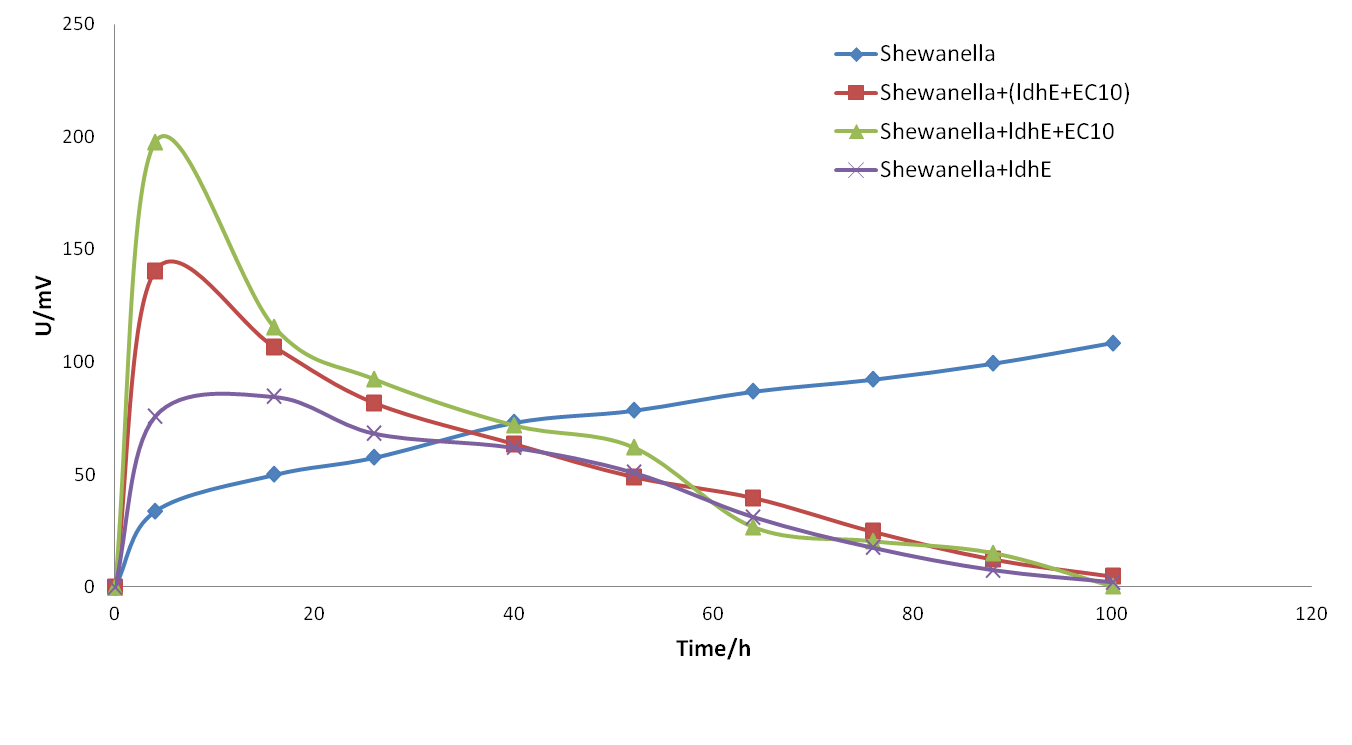
We established 5 groups of MFCs which contained Shewanella + lactate, Shewanella + LdhE, Shewanella + LdhE + B. subtilis, Shewanella + LdhE,Shewanella + LdhE + riboflavin and Shewanella + lactate + riboflavin respectively.

August 15th, 2015
Experiment 3: Solve low pH problem through adjusting bacteria proportion.
Firstly, we optimized the medium component by reducing ratio of C and N. Secondly, we optimized the proportion of the fermentation bacteria.
August 22th, 2015
Experiment 4: optimized the system continuously.

August 25th, 2015
Experiment 5: We first added Shewanella and then add other bacteria. Through this method, we achieved a higher but unstable voltage.

September 1st, 2015
Experiment 6: continuously optimize the system to achieve a stable voltage output.

September 7th, 2015
Experiment 7: change fermentation bacteria to B. subtilis.
Through many times experiment, we finally achieved our initial goal: a stable, high voltage output and long-term MFC!!!!

1 Molecular Manipulation
Our molecular cloning experiments go as follow:

During this process,we used the following protocols:
TransT1 transformation:
Protocol: 1. Mix 50 E.coli with 1 plasmids, and place them in ice cold water bath for 30 min. 2. Heat shock for 45 s at 42 ℃. Put the system back on ice for 2 min. 3. Add 500 μl of LB without antibiotics. Incubate at 37 ℃ for 30 min. The cultures are centrifuged for 1 h at 200 rpm. 4. Centrifuge the cultures at 4000 rpm.
Plasmids extraction
Protocol: 1. Harvest the bacterial cells by centrifugation at 12000 rpm for 1 min. 2. Add 250 μL buffer P1. 3. Add 250 μL buffer P2, turn the centrifugation tube up side down mildly for 6-8 times. 4. Add 350 μL buffer P3 and turn the up side down for 6-8 times immediately. Place it at 4 ℃ 5. Column equilibration: add 500 μL Buffer BL to the Spin Column CP3. Centrifuge for 1 min at 6. Centrifuge the centrifugation tube at 12000 rpm for 10 min. Pick 800 μL supernate and add them in the Spin Column CP3, and place it at 4 ℃temperature for 8-10 min. Then centrifuge the system at 12000 rpm for 1 min.Pour the liquid back to the column CP3. Repeat the former step. 7.Add 600 μl Buffer PW diluted with absolute ethanol into the column CP3 and centrifuge the system at 12000 rpm for 1 min. Repeat the former step. 8. Centrifuge the column CP3 at 12000 rpm for 2 min. Dry the column. 9. Add 50 μL EB buffer into the column CP3 and centrifuge the column at 12000 rpm for 2 min. Collect the plasmid in the liquid.
Restrictionenzyme digestion.
| Reaction system | 100 μL |
| Restrictionenzyme EcoRI | 5 μL |
| Restrictionenzyme RstI | 5 μL |
| 10 x buffer | 10 μL |
| H2O | 60μL |
Gel Extraction of DNA
1. Cut off the Single goal DNA band from the gel put it into the clean centrifuge tube and weigh. 2. Add same volume of Buffer PN more than the gel slice (0.1 g gel account for 100 μl). Incubate the mixture at 50 ℃ for 10 min until the gel has completely melted. 3.Place a column CA2 in a provided 2 ml collection tube. Add 500 μl Buffer BL to the CA2 column, and centrifuge at 12,000 rpm for 1 min at room temperature. Discard liquid and place the TIANGEN DNA column back into the same collection tube. 4. Place a column CA2 in a provided 2 ml collection tube. Add the mixture in last step to the column CA2, and place it at 4 ℃ temperature for 8-10 min,then centrifuge at 12,000 rpm for 1 min . Discard liquid and repeat the former step. 5. Add 600 μl Buffer PW into the column CA2 and centrifuge at 12,000 rpm for 1min. Pour out liquid and repeat the former step once. Attention: Buffer PW have to be diluted with absolute ethanol and store at 4 ℃. 6. Centrifuge at 12,000 rpm for 2 min. Dry the column. 7. Add 50 μl Buffer EB directly into the middle of the column membrane and place it at 60 ℃ temperature for 15 min. Centrifuge for 2 min at 12,000 rpm to elute DNA. An optional second elution will yield any residual DNA, though at a lower concentration.
Ligation DNA segments using pEasy-Blunt Cloning Kit.
Cloning system:
| ddH2O | pEasy vector | temperature | time | |
|---|---|---|---|---|
| mf-Lon 2 μL | 2 μL | 1 μL | 25 ℃ | 20 min |
| Rcfb-E 0.5 μL | 3.5 μL | 1 μL | 25 ℃ | 10 min |
PCR system:
1. PCR overlap System:
| 5 x Q5 buffer | 4 μL |
| dNTP | 1 μL |
| forwardprimer | 1 μL |
| reverseprimer | 1μL |
| DNA segments 1 | 0.3 μL |
| DNA segments 2 | 2.1 μL |
| Q5 enzyme | 0.2 μL |
| ddH2O | 10.4 μL |
2. Colony PCR system
| buffer | 25 μL |
| Forward primer | 5 μL |
| Reverse primer | 5 μL |
| dNTP | 10 μL |
| Taq | 5 μL |
| ddH2O | 200 μL |
3. PCR amplification system
| Buffer | 4.0 μL |
| dNTP | 2.0 μL |
| Template | 1.0 μL |
| Forward primer | 0.5 μL |
| Reverse primer | 0.5 μL |
| Taq | 0.5 μL |
| ddH2O | 11.5 μL |
PCR program:
Initial denaturation: 95 ℃ for 5 min 30cycles of: 95 ℃ for 30 min 55 ℃ for 30 min (different primers different annealing temperature) 72 ℃ for several min (“several”depends on the length of goal sequence, 1minper 1000bp) Final extension: 72 ℃ for 10 min
Tip: this program is just a “Simple PCR Program” that is only for reference. We need to adjust our protocol to adapt to different situations so we could get what we want.
Ligation
1. Check the concentration of DNA fragments and vector which are going to be ligated. 2. Calculate the amount of part A/partB and vector added, based on the fragment length. Note that a ligation using a molar ratio of 1:3 vector to inserts. 3. Add DNA/buffer and ligase together in the EP tube.
System:
| Part 1 | X μL |
| Vector | Y μL |
| 10 x T4 Ligase Buffer | 1.0 μL |
| T4 Ligase | 1.0 μL |
| ddH2O | Add until the total volume is 10.0 μL |
4. Mix the reaction by pipetting up and down gently and microfuge briefly. 5. Incubate at 22 ℃ for 1 hour or at 16 ℃ for overnight.
2 MFC set up
1. Preparation work.
1.1 Preparation of electrodes. We use carbon felt as the material of the electrodes which are cut to 2.5 cm X 2.5 cm dimension for anode and 2.5 cm X 3 cm (width X length) dimension for cathode. Steep the electrodes with acetone and 1 M hydrochloric acid for 24 hours and 12 hours respectively. After this step, electrodes are fastened to the copper wire with glue. The proton exchange membrane are steeped with 1 M hydrochloric acid overnight. 1.2 Assemble the chambers. Seal the assembled electrodes to the cover of the cell. Fix anode and cathode chambers together with clips and screw down the covers with electrodes. ATTENTION: DO NOT ASSEMBLE THE PROTON EXCHANGE MEMBRANE OR THEY WOULD MELT UNDER HIGH TEMPRATURE. 1.3 Sterilization. Autoclave to sterilize the devices and internal components at 121 ℃ for 20 min.
2. MFC construction.
2.1 Assemble the proton exchange membrane. 2.2 Fill the chambers with reaction system. Fill the anode chamber with anode solution. Pour 90 mL cathode medium (potassium ferricyanide) into cathode chamber. 2.3 Bacterial injection. 2.3.1 Calculate target OD600
Measure the OD600 in the flask and decide what OD600 you want in the chamber. The following equation will help you to determine how much bacterium solution you have to extract.
2.3.2 Bacterial injection Centrifuge the bacterium solution extracted from the flask and resuspend it with anode solution and inject them into the anode chamber. 2.4 Load a resistor into the circuit, and incubate the chamber at 37 ℃. The MFC is finished!
E-mail: ggjyliuyue@gmail.com |Address: Building No.20, No.92 Weijin road, Tianjin University, China | Zip-cod: 300072
Copyright 2015@TJU iGEM Team